
Abstract
The glycogen-accumulating organism (GAO) ‘Candidatus Competibacter’ (Competibacter) uses aerobically stored glycogen to enable anaerobic carbon uptake, which is subsequently stored as polyhydroxyalkanoates (PHAs). This biphasic metabolism is key for the Competibacter to survive under the cyclic anaerobic-‘feast’: aerobic-‘famine’ regime of enhanced biological phosphorus removal (EBPR) wastewater treatment systems. As they do not contribute to phosphorus (P) removal, but compete for resources with the polyphosphate-accumulating organisms (PAO), thought responsible for P removal, their proliferation theoretically reduces the EBPR capacity. In this study, two complete genomes from Competibacter were obtained from laboratory-scale enrichment reactors through metagenomics. Phylogenetic analysis identified the two genomes, ‘Candidatus Competibacter denitrificans’ and ‘Candidatus Contendobacter odensis’, as being affiliated with Competibacter-lineage subgroups 1 and 5, respectively. Both have genes for glycogen and PHA cycling and for the metabolism of volatile fatty acids. Marked differences were found in their potential for the Embden–Meyerhof–Parnas and Entner–Doudoroff glycolytic pathways, as well as for denitrification, nitrogen fixation, fermentation, trehalose synthesis and utilisation of glucose and lactate. Genetic comparison of P metabolism pathways with sequenced PAOs revealed the absence of the Pit phosphate transporter in the Competibacter-lineage genomes—identifying a key metabolic difference with the PAO physiology. These genomes are the first from any GAO organism and provide new insights into the complex interaction and niche competition between PAOs and GAOs in EBPR systems.
Similar content being viewed by others


Introduction
Enhanced biological phosphorus removal (EBPR) is a widely used process in wastewater treatment. It functions on the principle of creating favourable conditions for the enrichment of organisms capable of excess uptake and storage of phosphorus as polyphosphate. These organisms are collectively known as polyphosphate-accumulating organisms (PAOs). Selection of this phenotypic group is achieved by cycling biomass through carbon-rich anaerobic (‘feast’) and carbon-depleted aerobic (‘famine’) conditions—where heterotrophic organisms able to assimilate and store anaerobically available carbon are advantaged (Seviour et al., 2003). The PAO theoretically utilise aerobically stored polyphosphate to partly energise anaerobic carbon uptake. However, there are alternate non-PAO phenotypes suited to the feast: famine regime of EBPR. Such phenotypes have received considerable attention given that the organisms do not accumulate polyphosphate but compete with the PAO for substrates in the anaerobic feed stage, and therefore have a suggested negative influence on the P removal capacity of EBPR (Seviour et al., 2003; Oehmen et al., 2007).
One such phenotypic group comprises the glycogen-accumulating organisms (GAOs). These organisms theoretically energise anaerobic carbon uptake by the hydrolysis of aerobically stored glycogen with carbon storage as polyhydroxyalkanoates (PHAs). In the subsequent aerobic period, PHA stores are oxidised, providing energy and carbon for growth and the replenishment of glycogen stores (Liu et al., 1994). Two lineages of GAOs have been described, the alphaproteobacterial Defluviicoccus-related lineage and the gammaproteobacterial ‘Candidatus Competibacter phosphatis’ lineage (hereafter referred to as the Competibacter) (Crocetti et al., 2002; Wong et al., 2004; Oehmen et al., 2007). The Competibacter has an intragroup 16S rRNA gene identity of >89% and is thus much more diverse than a typical genus (approximately >93–95% (Clarridge, 2004)). Although limited phenotypic diversity is described, different Competibacter phylotypes are often found to coexist in EBPR systems (Kong et al., 2002, 2006). As with the Accumulibacter- (He et al., 2007) and Tetrasphaera-related PAO (Nguyen et al., 2011) and the Defluviicoccus-related GAO (Burow et al., 2007; McIlroy and Seviour, 2009), the composition of Competibacter communities differs between, and temporally within, EBPR systems (Kong et al., 2006; Wong and Liu, 2006), indicating niche differentiation. Thus, resource competition between the PAOs and GAOs is likely to occur across a range of niches in complex full-scale systems.
Given the absence of cultured representatives of the Competibacter, our understanding of the physiology and ecology of GAO lineages is based on in situ microautoradiography (MAR) studies and metabolic modelling of bulk biochemical transformations in enriched cultures (Oehmen et al., 2007). These approaches have provided valuable information but are somewhat limited in the physiological detail they can provide. In addition, metabolic models are based on enrichment studies, where the composition of the Competibacter-lineage community is rarely defined. The presence of multiple species in these enrichment cultures may explain apparently conflicting evidence for the metabolism of glycogen, with separate studies suggesting the operation of either the Embden–Meyerhof–Parnas (EMP) or the Entner–Doudoroff (ED) glycolytic pathway (Oehmen et al., 2010).
A ground-breaking study in microbial ecology research was the assembly of the genome for a member of the thus-far unculturable PAO ‘Candidatus Accumulibacter phosphatis’ (henceforth referred to as Accumulibacter) through metagenomics (García-Martín et al., 2006). This allowed construction of a detailed metabolic pathway model and provided the basis for transcriptomic and metaproteomic studies (Wilmes et al., 2008; Wexler et al., 2009; He et al., 2010), together substantially advancing our understanding of the ecology of Accumulibacter (He and McMahon, 2011). More recently, omic methods have allowed the assembly of genomes representing as low as 0.1% of a metagenome (Wrighton et al., 2012; Albertsen et al., 2013), thus circumventing the need to either culture or enrich for the target group for genome assembly.
In this study we used these new metagenomic methods to assemble two Competibacter genomes, representing distinct species, from two short-term enrichment cultures. In addition, their in situ abundance and diversity were investigated by sequencing a metagenome from the biomass of the full-scale wastewater treatment plant used to seed both enrichment reactors. This study is the first of its kind for a GAO organism and resolves some of the important unanswered questions regarding GAO physiology, such as glycolysis pathways and denitrification ability. Comparison with genomic-based pathway models for the Accumulibacter (García-Martín et al., 2006) and Tetrasphaera PAOs (Kristiansen et al., 2013) gives further insight into the genetic bases of the PAO phenotype.
Materials and methods
Samples and sequencing batch reactor operation
An activated sludge sample was collected from the Ejby Mølle wastewater treatment plant, Odense, Denmark (55.398487, 10.420596), and used for inoculation of a 4.3l sequencing batch reactor with anaerobic (feast): aerobic (famine) cycling using acetate as the sole carbon source (see Supplementary Information for details). Samples from day 89 of ‘Run B’ were used for the metagenomic sequencing presented in this paper. An additional Competibacter genome, extracted from a metagenome of a sample from day 39 of a separate enrichment (Run A) (Supplementary Information) (Albertsen et al., 2013), was also analysed in this study. The full-scale activated sludge sample for metagenomics was taken from the aeration tank at the Ejby Mølle wastewater treatment plant on 8 November 2011. Samples for seeding the enrichment reactor for Runs A and B were taken from the same plant on 19 October 2011 and 11 November 2010, respectively. Biochemical transformations were monitored as described in Supplementary Information and a typical profile is given in Supplementary Figure S1. The microbial community was also monitored in situ with fluorescent in situ hybridization (FISH) (Daims et al., 2005), and PHA storage in cells was visualised after Nile Blue A staining (Ostle and Holt, 1982) (data not shown).
DNA extraction, sequencing library preparation and sequencing
DNA was extracted from 1 ml samples using the FastDNA spin kit for soil (MP Biomedicals, Solon, OH, USA) according to the manufacturer’s instructions but with an initial incubation for 3 min in phenol at 90 °C (see Supplementary Information for further details).
The enrichment sample was prepared for sequencing using the TruSeq DNA Sample Preparation Kit v2 (Illumina Inc., San Diego, CA, USA) with 2 μg of DNA following the manufacturer’s instructions with nebuliser fragmentation. The full-scale sample was prepared using the Nextera kit (Illumina Inc.) according to the manufacturer’s instructions. Library DNA concentration was measured using the QuantIT kit (Molecular Probes, Life Technologies, Naerum, Denmark) and paired-end sequenced (2 × 150 bp) on an Illumina HiSeq2000 using the TruSeq PE Cluster Kit v3-cBot-HS and TruSeq SBS kit v.3-HS sequencing kit (Illumina Inc.).
Metagenome assembly
Metagenome reads in fastq format were imported to CLC Genomics Workbench v. 5.1 (CLC Bio, Aarhus, Denmark) and trimmed using a minimum phred score of 20, a minimum length of 50 bp, allowing no ambiguous nucleotides and trimming off Illumina sequencing adaptors if found. The trimmed metagenome reads were assembled using CLC’s de novo assembly algorithm, using a kmer of 63 and a minimum scaffold length of 1 kbp.
Genome binning, completeness estimation and finishing
For genome binning, a combination of coverage and tetranucleotide frequency patterns was used as described previously (Albertsen et al., 2013). Genome coverage was estimated by mapping the metagenome reads to the assembled scaffolds using the CLC’s map reads to a reference algorithm with a minimum similarity of 95% over 100% of the read length. Tetranucleotide frequency patterns were calculated for all scaffolds and imported to R (http://www.R-project.org) for principal component analysis using the Vegan package version 2.0-5 (Oksanen et al., 2011).
Open reading frames were predicted in the assembled scaffolds using FragGeneScan (Rho et al., 2010) with the following settings: complete=1 train=complete. A set of 105 Hidden Markov Models (HMM) of essential proteins (Dupont et al., 2012) conserved in all Gammaproteobacteria were searched against the predicted open reading frames using HMMER3 (http://hmmer.org) with the default settings, except that the trusted cutoff was used (-cut_tc). The identified proteins were taxonomically classified using blastp against the RefSeq protein database (version 52) with a maximum e-value cutoff of 1e-5. MEGAN (Huson et al., 2011) was used to extract consensus taxonomic level assignments from the blast xml output file.
The complete approach to extract individual genomes from metagenomes is described in detail in the online guide http://madsalbertsen.github.io/multi-metagenome/ (Albertsen et al., 2013).
The genome sequence data have been submitted to DDBJ/EMBL/GenBank databases under accession numbers CBTJ020000001-CBTJ0000120 (Run A) and CBTK010000001-CBTK010000319 (Run B), and the raw metagenome reads have been deposited in NCBI SRA under accession numbers SRR628916 (Run B) and SRR999554 (full-scale).
Genome annotation and metabolic reconstruction
Contigs for each genome were uploaded to the ‘MicroScope’ annotation pipeline (Vallenet et al., 2006, 2009). Automatic annotations were validated manually for the genes involved in metabolic pathways of interest with the assistance of the integrated MicroCyc and KEGG (Kyoto Encyclopedia of Genes and Genomes) databases.
Comparative genomics
Genome-wide similarity between the two Competibacter genomes was calculated using Jspecies software (IMEDEA, Esporles, Spain) (Richter and Rosselló-Móra, 2009) with the default BLAST implementation.
Estimation of in situ abundance and diversity
Metagenome reads from the full-scale sample were mapped to the two Competibacter genomes requiring at least 75% similarity using CLC’s reference mapping algorithm and exported in SAM format. A custom perl script was used to convert the SAM file into a format that could be visualised in R. All utilised scripts can be found at http://madsalbertsen.github.io/multi-metagenome/.
Results and discussion
The metagenome sequencing of the enrichment culture resulted in 30 Gbp of sequence after quality filtering. Using a combination of coverage and tetranucleotide frequency, it was possible to extract a Competibacter genome (Figure 1 and Table 1—Run B). In addition, another Competibacter genome was available from a previous experiment on another enrichment reactor (Table 1—Run A) (Albertsen et al., 2013). Although the two genomes are in 112 and 319 contigs, respectively, they can probably be considered effectively complete due to the presence of all essential genes. The fragmentation is mainly the result of repeat elements >750 bp that could not be spanned by the paired-end reads given the insert size of ∼300 bp. The two genomes have an average nucleotide identity of 74% over the ∼40% of the genome aligned (1.5 Mbp), indicating that they belong to separate species (>95% species cutoff (Richter and Rosselló-Móra, 2009)).

Extraction of the Competibacter-lineage genome from the metagenome (Run B) using coverage and PCA on tetranucleotide frequencies. Each circle represents a metagenomic scaffold, with size proportional to length and coloured by GC content. Only metagenome scaffolds >10 kbp are shown. The box encloses fragments representing the Competibacter lineage.
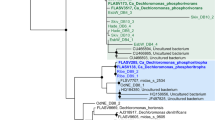
Phylogeny of the Competibacter species
Phylogenetic analysis of the 16S rRNA genes, representing the assembled Competibacter genomes from Runs A and B of the enrichment reactors, indicates that they are associated with the previously defined subgroups 1 and 5 (Kong et al., 2002), respectively (Figure 2). In general, the Competibacter lineage is much more diverse than originally defined (see Figure 2) and has substantially expanded since the original description of ‘Candidatus Competibacter phosphatis’ (Crocetti et al., 2002), the sequences of which now make up subgroup 3 (Figure 2). In situ studies applying the available Competibacter-lineage subgroup probes reveal few phenotypic differences, with all phylotypes adhering to the basic GAO phenotype in their ability to anaerobically store volatile fatty acids (VFAs) as PHA without observed polyphosphate cycling (Kong et al., 2006). However, important phenotypic differences are likely, supported by the common co-existence of subgroups, which needs to be considered when interpreting results using the commonly applied broad group probes (GAOmix (Crocetti et al., 2002), GB, GBmix (Kong et al., 2002) and GB742 (Kim et al., 2011)). It is also possible that some phylogenetic clades (Figure 2), for which no in situ data are currently available, do not behave according to the GAO phenotype. In addition, given that sequence similarity among the members is >89%, we believe it more appropriate to refer to the Competibacter group as the Competibacteraceae family, rather than inferring it to be a single species or genus as in the past. This family is also suggested to include the related genus Plasticicumulans, which is also shown to cycle PHA in feast–famine-regime-activated sludge systems (Schroeder et al., 2009; Jiang et al., 2011). On the basis of our phylogenetic analysis (Supplementary Information; Figure 2), for the Run A (subgroup 1) genome, we suggest the provisional name ‘Candidatus Competibacter denitrificans’ (C. denitrificans), based on its annotated ability for denitrification (see later). For the Run B (subgroup 5-related) genome, we suggest the name ‘Candidatus Contendobacter odensis’ (C. odensis) based on the location of the seed sludge from which it originated (Odense, Denmark) and the novel genus name maintaining a theme reflecting the importance of the family as competitors of the PAOs (Latin: Contendo—‘compete’).

Maximum-likelihood (PhyML) phylogenetic tree of all Competibacter-lineage-related 16S rRNA gene sequences (see Supplementary Information). Non-parametric bootstrap (from 100 analyses) branch-support values >50% are included. The inner bracket on the right indicates sequences within the currently defined Competibacter lineage and divisions indicate previous probe-defined subgroups (Kong et al., 2002; Kim et al., 2011). *Denotes clusters in which the majority of sequences are sourced from activated sludge systems. The scale bar represents substitutions per nucleotide base.
Revision of the metabolic model for the Competibacteraceae through genomics
Much of the current understanding of GAO metabolism largely comes from mass biochemical transformations of enriched cultures, coupled with ‘selective’ inhibition of key enzymes, with subsequent extrapolation to predict the metabolic pathways involved. The two Competibacteraceae genomes allow direct insight into the genetic potential for the metabolic pathways that are critical for understanding the competition between GAOs and PAOs. Curated annotations for selected pathways are given in Supplementary Table S1, and expanded and simplified diagrammatic metabolic models are presented in Supplementary Figures S2 and Figure 3, respectively. Key differences in metabolic potential are compared in Table 2.

Diagrammatic representation of the metabolic pathways for the Competibacteraceae species in EBPR-activated sludge treatment plants (a) in the absence of electron acceptors (anaerobic) and (b) in the presence of the electron acceptors oxygen (aerobic) or nitrate. ETC=electron transport chain; PEP=phosphoenolpyruvate; KDPG=2-dehydro-3-deoxy-D-gluconate-6-phosphate; TAG=triacylglycerols; PHB=polyhydroxybutyrate; PHV=polyhydroxyvalerate; PH2MB=polyhydroxy-2-methylbutyrate; and PH2MV=polyhydroxy-2-methylvalerate. For more details on the pathways please refer to Supplementary Figure S2 in conjunction with Supplementary Table S1.
Anaerobic carbon uptake in Competibacteraceae
In wastewater treatment plants, PAOs and GAOs compete for the by-products of hydrolysis and fermentation of more complex organic material, such as simple sugars, lactate, acetate, and propionate (Nielsen et al., 2010). Mathematical models of the GAO phenotype consider acetate and propionate given their relative high abundance in EBPR systems (Oehmen et al., 2007), and both Competibacteraceae genomes have the genes necessary for the uptake and utilisation of these substrates. In addition, lactate can be used aerobically by both organisms, although only C. denitrificans appear to be able to utilise lactate anaerobically (Supplementary Information).
Both MAR-FISH and enrichment cultures confirm the uptake of acetate and propionate by Competibacteraceae, although propionate consumption is relatively less efficient (Oehmen et al., 2004; Kong et al., 2006). Such a difference was not observed in Accumulibacter enrichments (Oehmen et al., 2005), and these differences have been exploited to reduce Competibacteraceae competitiveness in mixed laboratory-scale cultures (Lu et al., 2006). In the absence of observed genetic differences in the genes with predicted involvement in VFA metabolism (Supplementary Information), such an advantage of Accumulibacter is probably due to differences in transport affinity or regulation, also keeping in mind that propionate uptake efficiency may vary between strains (Seviour and McIlroy, 2008).
The acetate/propionate permease (actP) annotated in the Competibacteraceae genomes (Supplementary Table S1), and Accumulibacter (García-Martín et al., 2006), facilitates the transport of acetate and propionate in symport with a cation. Saunders et al. (2007) hypothesised that VFA uptake in the Competibacteraceae is energised by a proton motive force (PMF) generated by the export of protons by an F1F0-ATPase at the expense of ATP, based on the observed significant reduction in acetate uptake in the presence of the membrane ATPase inhibitor, N-N’-dicyclohexylcarboiimide. The genes for such an ATPase transporter complex are located in both genomes. In addition, the export of protons by the action of a fumarate reductase complex was also predicted to contribute to the PMF (see later). The location of a possible sodium-export carboxylase, with homology to methylmalonyl-CoA decarboxylase complex genes in other organisms (mmdABC (Bott et al., 1997); Supplementary Information), in both genomes also suggests the possible involvement of a sodium potential in VFA uptake, as has been suggested for the Defluviicoccus-related GAO based on inhibitor studies in enriched cultures (Burow et al., 2008).
Anaerobic glycogen metabolism
The defining attribute of the GAO metabolism in EBPR is the use of glycogen stores to supply the energy and reducing equivalents for the uptake and storage of carbon. Whether the EMP or the ED pathway is utilised has substantial impact on the redox balance of the cell, given the two alternate routes generate three and two moles ATP per mole of glucose-6-phosphate monomer, respectively. Thus, the less energy efficient ED pathway gives predicted higher glycogen utilisation and PHA production under anaerobic conditions (Oehmen et al., 2010). Both genomes analysed here have genes for the complete EMP pathway, whereas the ED pathway was only identified in the C. denitrificans genome. The possible ability of the C. denitrificans to switch between the two pathways would give increased flexibility in the anaerobic balance of the redox potential of the cell. However, the oxidative pentose phosphate pathway genes encoding enzymes that can convert the products of glycogen catabolism to 6-phosphogluconate, the beginning of the ED pathway, were not detected in either genome. Alternative routes to the pentose phosphate pathway are possible, but not evident in the genome, and it is therefore unclear whether the ED can be used to metabolise glucose originating from glycogen. In other organisms possessing both the ED and EMP pathways, the ED pathway is induced in the presence of specific carbon sources, with the EMP pathway forming the core catabolic route (Conway, 1992), with similar roles for these alternate pathways therefore possible for C. denitrificans.
The EMP pathway is included in the current metabolic models for Competibacteraceae, which fit empirical enrichment culture data well (Oehmen et al., 2010). However, evidence at higher temperatures (30 °C) (Lemos et al., 2007; Lopez-Vazquez et al., 2009), albeit inconsistently (Bengtsson, 2009), suggests the operation of the ED pathway in Competibacteraceae enrichments. On the basis of these observations it has been suggested that either individual Competibacteraceae species shift their reliance on the EMP to the ED pathway in response to certain conditions (that is, increased temperature) or such conditions select for distinct species with different metabolic potentials for glycogen catabolism pathways (Oehmen et al., 2010). Ultimately, a study using genomics (and perhaps transcriptomics and/or proteomics) analysis of a culture that has demonstrated the stoichiometry of the ED pathway will be required to answer these critical questions, but the data presented here importantly show that different Competibacteraceae species can vary in their genetic potential for glycolytic pathways.
Carbon storage and redox balance in the absence of an external electron acceptor
The ability to store anaerobically exogenous carbon as PHA is the key to the competitiveness of Competibacteraceae under dynamic EBPR conditions. The reducing equivalents required for polymerisation of VFA-coenzyme A ester moieties are predicted to result from glycolysis (Oehmen et al., 2007). However, in the absence of polyphosphate as a major energy source, relative to the PAO metabolism, more glycogen hydrolysis is required to meet the anaerobic energy demand. Thus, reducing equivalents are produced in excess of what is required for PHA synthesis. This excess is theoretically balanced with the flux of pyruvate through the reductive branch of the tricarboxylic acid cycle and the succinate-propionate pathway to propionyl-CoA (Mino et al., 1998). This ester is in turn condensed with acetyl-CoA to form polyhydroxyvalerate and is evidenced by the production of both polyhydroxybutyrate and polyhydroxyvalerate from acetate in GAO enrichments (Oehmen et al., 2007). Fumarate reductase activity is key to the operation of the reductive branch of the tricarboxylic acid cycle and is also predicted to contribute to the PMF required for acetate uptake (Saunders et al., 2007). One of the two sets of genes for the succinate/fumarate reductase complex, annotated in each Competibacteraceae genome, likely serves this function, although differentiation between the reaction direction catalysed by each complex needs to be empirically determined (Cecchini et al., 2002).
Interestingly, C. denitrificans alone has the genetic potential for fermentation of glucose-6P to lactate, as well as the ability to assimilate glucose, which could also contribute to the utilisation of excess reducing power. Ability to utilise glucose has never been directly observed in any of the Competibacteriaceae phylotypes (Kong et al., 2006), although they have been found in abundance in glucose-fed EBPR reactors (Zengin et al., 2010; Begum and Batista, 2012). Glucose assimilation was therefore assessed in this study using MAR-FISH analysis of activated sludge from the Ejby Mølle wastewater treatment plant used to seed the enrichment reactors (Supplementary Information). This analysis revealed that essentially all the observed Competibacteraceae cells assimilated glucose under both aerobic and anaerobic conditions (Supplementary Figure S3), thus supporting the ability for glucose assimilation and fermentation that is suggested by annotation of the C. denitrificans genome. The ability to ferment glucose and store PHA is shared with the Tetrasphaera japonica PAO (Kristiansen et al., 2013) and the Defluviicoccus vanus GAO (Wong and Liu, 2007). Interestingly, anaerobic energy demand for C. denitrificans may be supplemented by the excretion of lactate, produced from glycogen fermentation, in symport with a cation contributing to a PMF (Konings et al., 1984). The importance of fermentation, along with alternative pathways to balance the anaerobic reducing power, has been discussed elsewhere (Supplementary Information).
Anaerobic storage polymers
Both subgroup genomes contain the genes for the synthesis and hydrolysis of PHAs. These include multiple PHA synthases, the key enzyme catalysing the ultimate step in PHA synthesis (Rehm, 2003). In addition, C. odensis also contains a diacylglycerol O-acyltransferase (EC 2.3.1.20), which catalyses the ultimate step in triacylglycerol and waxy ester synthesis (Kalscheuer and Steinbüchel, 2003). The gene was also annotated in the Accumulibacter clade IIA genome, although neither triacylglycerol nor wax ester production was detected in a lab-scale EBPR community containing Accumulibacter (Wexler et al., 2009). Nevertheless, this does not preclude a possible role for triacylglycerol synthesis in some Competibacteraceae or Accumulibacter species in the more complex full-scale systems, or in their natural environment (Supplementary Information).
Aerobic metabolism of Competibacteraceae
Under the carbon-deficient aerobic conditions of EBPR, Competibacteraceae is predicted to mobilise stored PHA for growth and replenishment of glycogen stores. The genomic potential for the glyoxylate shunt (Kornberg and Krebs, 1957), and possibly also the anaplerotic ethylmalonyl-CoA pathway (Erb et al., 2007,2009) (Supplementary Information), allows growth on acetyl-CoA generating compounds such as PHAs.
Both subgroups contain the genes for the classical pathway for glycogen synthesis (glgABC) along with those for its hydrolysis to its glucose moieties (glgPX) (Preiss, 2006). Glycogen synthesis is normally associated with growth-limiting conditions, where growth has slowed down or ceased (Preiss, 2006). For Competibacteraceae in the EBPR environment, this probably reflects the limitation of growth nutrients relative to the carbon available from stored PHA. However, in Mycobacterium smegmatis glycogen cycling is also suggested to occur during exponential growth, where the compound acts as a carbon capacitor for glycolysis. Carbon not immediately required is shuttled into glycogen and then accessed when needed during growth (Belanger and Hatfull, 1999). Such aerobic cycling of glycogen with simultaneous operation of the ED and gluconeogenesis pathways was also suggested in a nuclear magnetic resonance-based study with a Competibacteraceae enrichment (Lemos et al., 2007). The benefit of aerobic cycling of carbon through the ED pathway is unclear, but in other organisms it is associated with the maintenance of intermediates of polysaccharide synthesis pathways, principally fructose-6-phosphate (Portais and Delort, 2002). Supporting this possible role for the ED pathway in C. denitrificans is the colocalisation of the eda gene, a key to the ED pathway, with genes involved in glucuronate metabolism. Glucuronate is a component of the characterised exopolysaccharide ‘granulan’ (Seviour et al., 2010) whose synthesis is linked to the Competibacteraceae (Seviour et al., 2011).
Interestingly, annotation of the genomes reveals capabilities to synthesise the non-reducing disaccharide trehalose in the C. odensis genome only (Supplementary Information). Three separate routes were found: the OtsAB-, TreS- and TreXYZ-mediated pathways, which enable trehalose synthesis from glucose, maltose and glycogen, respectively (Chandra et al., 2011). Trehalose has a number of proposed functions in Bacteria, including imparting resistance to desiccation, relative extremes in temperature, and oxidative and osmotic stresses (Elbein et al., 2003). The direct links between trehalose and glycogen also provide a potential buffering system against adverse environmental conditions, such as salinity, even in the absence of an exogenous carbon supply (Wolf et al., 2003). Thus, a mixture of both trehalose and glycogen may act as carbon storage reservoirs in C. odensis with the environmental conditions determining their ratio. Such a metabolic feature would be missed by most studies given that glycogen storage is rarely measured directly and is instead inferred from quantification of the total cellular carbohydrates (Smolders et al., 1994), which would include trehalose.
Polyphosphate metabolism
Polyphosphate cycling is not perceived to have a central role in the metabolism of the Competibacteraceae. However, a genetic comparison of the pathways involved in PAOs gives an insight into the potential basis for the PAO phenotype. Currently, representative genomes are available for three putative PAOs: Accumulibacter (García-Martín et al., 2006), Tetrasphaera (Kristiansen et al., 2013) and Microlunatus phosphovorus (Kawakoshi et al., 2012).
Given that polyphosphate synthesis has been detected in all species tested (Brown and Kornberg, 2008), it is not surprising that the ability to synthesise and hydrolyse polyphosphate is also possessed by organisms with a GAO phenotype (see Supplementary Information, Supplementary Table S2 and Supplementary Figure S4). The presence of polyphosphate in GAO cells has also been seen from the staining analysis of some Competibacteraceae species in situ (Nielsen et al., 1999).
The key observable difference between the PAO and GAO appears to be in the phosphate transport systems. The two Competibacteraceae species contain putative genes encoding for the high-affinity Pst uptake system only, and not the low-affinity Pit system (Kornberg et al., 1999) as in the PAOs Accumulibacter, Tetrasphaera and M. phosphovorus. The apparent functional redundancy in PAOs is postulated to allow uptake throughout the aerobic phase, irrespective of exogenous inorganic phosphorus concentrations (García-Martín et al., 2006). The key role for the Pit system in PAOs is in the generation of a PMF under anaerobic conditions, generated by the export of metal-phosphates in symport with a proton (van Veen et al., 1994), which seems to drive VFA uptake in Accumulibacter (Saunders et al., 2007). The Pit system is also absent in ‘Candidatus Microthrix parvicella’ (McIlroy et al., 2013), which accumulate excess polyphosphate but do not perform P cycling that is linked to anaerobic carbon uptake (Andreasen and Nielsen, 2000). Although additional regulatory genes likely contribute to the PAO phenotype in EBPR, the Pit system appears to be a prerequisite for such a metabolism.
Nitrogen metabolism
The Competibacteraceae species show substantial differences in the pathways they possess for nitrogen-related metabolism (Table 2). Competibacteraceae species are reported to have varied abilities for denitrification in situ (Kong et al., 2006). Denitrifying ability for some Competibacteraceae is also suggested by the biochemical transformations of enrichment cultures (Zeng et al., 2003; Wang et al., 2008). The two genomes studied here demonstrate such variation, with only C. denitrificans possessing the genetic potential for denitrification from nitrate to nitrogen (see Supplementary Information). Although C. odensis also possesses the ability of nitrate reduction to nitrite, it possesses an assimilatory nitrite reductase, and thus instead possesses the metabolic potential to reduce nitrate to ammonia via nitrite. An additional difference in the nitrogen metabolism of the Competibacteraceae species is the ability of C. odensis to fix nitrogen. Such an ability is perhaps not relevant to the nitrogen-rich-activated sludge environment, but likely reflects adaptation to its source environment as suggested for the Accumulibacter clade IIA strain (García-Martín et al., 2006) (see Supplementary Information).
Similarity to full-scale in situ strains
The use of laboratory-scale enrichments simplifies the assembly of genomes by lowering the complexity of the system (Albertsen et al., 2013). However, there is the possibility that lab-scale enrichments do not promote the growth of the abundant in situ strains. Metagenomics enables direct comparison of the enrichment genomes with the in situ strains, thereby indicating the relevance of the annotated genomes. Therefore, a metagenome was established from the seeding full-scale Ejby Mølle EBPR wastewater treatment plant (127 million reads, 15 Gbp). The reads were used to estimate the in situ abundance and similarity to the two enrichment genomes. The genome of C. denitrificans was identical to an in situ Competibacteraceae genome with a metagenome abundance of 1% (FISH estimated bio-volume abundance was determined to be 3–4% for Competibacteraceae in the treatment plant (Mielczarek et al., 2013)). No abundant in situ strains were closely related to the C. odensis genome. Given the ability of C. denitrificans to denitrify and possibly ferment, this species appears relatively better suited to the dynamic availability of electron acceptors in the EBPR environment, which is consistent with its detection as an abundant species in the full-scale-derived metagenome. Interestingly, closer inspection of the full-scale metagenome revealed that there appeared to be an additional abundant Competibacteraceae genome at ∼1% metagenome abundance and several very low abundant strains (<0.1% metagenome abundance). It was possible to reconstruct a partial Competibacteraceae genome from the full-scale plant, which revealed that the second abundant in situ Competibacteraceae was <75% similar at the genome level (average nucleotide identity) to both enrichment genomes, thereby clearly representing another species. This supports earlier observations that a number of different Competibacteraceae species coexist in full-scale EBPR plants.
Perspectives
The primary selection pressure of the EBPR systems is the creation of anaerobic feast followed by famine in the presence of external electron acceptors. Thus, the ability to anaerobically store carbon as PHA for later use appears to be the principal selection pressure for the Competibacteraceae population species. Features showing less evolutionary conservation in the lineage appear to relate to potential pathways for the anaerobic source of energy and redox balance, nitrogen and exopolysaccharide sugar-related metabolism (Table 2), and could be considered to constitute ‘secondary’ selection pressures that relate to niche partitioning. Such differences are suggested for the different strains of the Accumulibacter (He and McMahon, 2011) and likely give these populations combined metabolic flexibility and subsequent stability. Consequently, establishing relevant phenotypic divisions within the lineages needs further investigation and consideration when metabolic models of the complex interaction between PAO and GAO populations are devised. Continued attempts should be made to culture these organisms, and the genome information here may provide clues to achieve this. However, in providing the first information on the genetic potential of a putative GAO, this study is an important initial step in elucidating the details of their in situ physiology, providing a foundation for future transcriptomic and proteomic studies.
Accession codes
Accessions
GenBank/EMBL/DDBJ
References
Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, Nielsen PH . (2013). Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol 31: 533–538.
Andreasen K, Nielsen PH . (2000). Growth of Microthrix parvicella in nutrient removal activated sludge plants: studies of in situ physiology. Water Res 34: 1559–1569.
Begum SA, Batista JR . (2012). Microbial selection on enhanced biological phosphorus removal systems fed exclusively with glucose. World J Microbiol Biotechnol 28: 2181–2193.
Belanger AE, Hatfull GF . (1999). Exponential-phase glycogen recycling is essential for growth of Mycobacterium smegmatis. J Bacteriol 181: 6670–6678.
Bengtsson S . (2009). The utilization of glycogen accumulating organisms for mixed culture production of polyhydroxyalkanoates. Biotechnol Bioeng 104: 698–708.
Bott M, Pfister K, Burda P, Kalbermatter O, Woehlke G, Dimroth P . (1997). Methylmalonyl-CoA decarboxylase from Propionigenium modestum—cloning and sequencing of the structural genes and purification of the enzyme complex. Eur J Biochem 250: 590–599.
Brown MR, Kornberg A . (2008). The long and short of it - polyphosphate, PPK and bacterial survival. Trends Biochem Sci 33: 284–290.
Burow L, Mabbett A, McEwan A, Bond P, Blackall L . (2008). Bioenergetic models for acetate and phosphate transport in bacteria important in enhanced biological phosphorus removal. Environ Microbiol 10: 87–98.
Burow LC, Kong Y, Nielsen JL, Blackall LL, Nielsen PH . (2007). Abundance and ecophysiology of Defluviicoccus spp., glycogen-accumulating organisms in full-scale wastewater treatment processes. Microbiology 153: 178–185.
Cecchini G, Schroder I, Gunsalus RP, Maklashina E . (2002). Succinate dehydrogenase and fumarate reductase from Escherichia coli. Biochim Biophys Acta 1553: 140–157.
Chandra G, Chater KF, Bornemann S . (2011). Unexpected and widespread connections between bacterial glycogen and trehalose metabolism. Microbiology 157: 1565–1572.
Clarridge III JE . (2004). Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin Microbiol Rev 17: 840–862.
Conway T . (1992). The Entner-Douderoff pathway: history, physiology and molecular biology. FEMS Microbiol Rev 103: 1–28.
Crocetti GR, Banfield JF, Keller J, Bond PL, Blackall LL . (2002). Glycogen-accumulating organisms in laboratory-scale and full-scale wastewater treatment processes. Microbiology 148: 3353–3364.
Daims H, Stoecker K, Wagner M . (2005). Fluorescence in situ hybridization for the detection of prokaryotes. In: Molecular Microbial Ecology. Taylor & Francis: New York, NY, USA, pp 213–239.
Dupont CL, Rusch DB, Yooseph S, Lombardo MJ, Richter RA, Valas R et al (2012). Genomic insights to SAR86, an abundant and uncultivated marine bacterial lineage. ISME J 6: 1186–1199.
Elbein AD, Pan YT, Pastuszak I, Carroll D . (2003). New insights on trehalose: a multifunctional molecule. Glycobiology 13: 17R–27R.
Erb TJ, Berg IA, Brecht V, Muller M, Fuchs G, Alber BE . (2007). Synthesis of C5-dicarboxylic acids from C2-units involving crotonyl-CoA carboxylase/reductase: the ethylmalonyl-CoA pathway. Proc Natl Acad Sci U S A 104: 10631–10636.
Erb TJ, Fuchs G, Alber BE . (2009). (2S)-Methylsuccinyl-CoA dehydrogenase closes the ethylmalonyl-CoA pathway for acetyl-CoA assimilation. Mol Microbiol 73: 992–1008.
García-Martín H, Ivanova N, Kunin V, Warnecke F, Barry K, McHardy A et al (2006). Metagenomic analysis of two enhanced biological phosphorus removal (EBPR) sludge communities. Nature Biotechnol 24: 1263–1269.
He S, McMahon KD . (2011). Microbiology of ‘Candidatus Accumulibacter' in activated sludge. Microbial Biotechnol 4: 603–619.
He S, Kunin V, Haynes M, Martin HG, Ivanova N, Rohwer F et al (2010). Metatranscriptomic array analysis of 'Candidatus Accumulibacter phosphatis'-enriched enhanced biological phosphorus removal sludge. Environ Microbiol 12: 1205–1217.
He S, Gall DL, McMahon KD . (2007). "Candidatus Accumulibacter" population structure in enhanced biological phosphorus removal sludges as revealed by polyphosphate kinase genes. Appl Environ Microbiol 73: 5865–5874.
Huson DH, Mitra S, Ruscheweyh HJ, Weber N, Schuster SC . (2011). Integrative analysis of environmental sequences using MEGAN4. Genome Res 21: 1552–1560.
Jiang Y, Sorokin DY, Kleerebezem R, Muyzer G, van Loosdrecht M . (2011). Plasticicumulans acidivorans gen. nov., sp. nov., a polyhydroxyalkanoate-accumulating gammaproteobacterium from a sequencing-batch bioreactor. Int J Syst Evol Microbiol 61: 2314–2319.
Kalscheuer R, Steinbüchel A . (2003). A novel bifunctional wax ester synthase/acyl-CoA:diacylglycerol acyltransferase mediates wax ester and triacylglycerol biosynthesis in Acinetobacter calcoaceticus ADP1. J Biol Chem 278: 8075–8082.
Kawakoshi A, Nakazawa H, Fukada J, Sasagawa M, Katano Y, Nakamura S et al (2012). Deciphering the genome of polyphosphate accumulating actinobacterium Microlunatus phosphovorus. DNA Res 19: 383–394.
Kim JM, Lee HJ, Lee DS, Lee K, Jeon CO . (2011). Identification of a novel subgroup of uncultured gammaproteobacterial glycogen-accumulating organisms in enhanced biological phosphorus removal sludge. Microbiology 157: 2287–2296.
Kong Y, Xia Y, Nielsen JL, Nielsen PH . (2006). Ecophysiology of a group of uncultured gammaproteobacterial glycogen-accumulating organisms in full-scale enhanced biological phosphorus removal wastewater treatment plants. Environ Microbiol 8: 479–489.
Kong Y, Ong SL, Ng WJ, Liu W . (2002). Diversity and distribution of a deeply branched novel proteobacterial group found in anaerobic-aerobic activated sludge processes. Environ Microbiol 4: 753–757.
Konings WN, Otto R, Ten Brink B, Robillard GT, Elferink MG, Hellingwerf KJ . (1984). Relation between the protonmotive force and solute transport in bacteria. Biochem Soc Trans 12: 152–154.
Kornberg HL, Krebs HA . (1957). Synthesis of cell constituents from C2-units by a modified tricarboxylic acid cycle. Nature 179: 988–991.
Kornberg A, Rao NN, Ault-Riché D . (1999). Inorganic polyphosphate: a molecule of many functions. Annu Rev Biochem 68: 89–125.
Kristiansen R, Nguyen HTT, Saunders AM, Nielsen JL, Wimmer R, Le VQ et al (2013). A metabolic model for members of the genus Tetrasphaera involved in enhanced biological phosphorus removal. ISME J 7: 543–554.
Lemos P, Dai Y, Yuan Z, Keller J, Santos H, Reis M . (2007). Elucidation of metabolic pathways in glycogen-accumulating organisms with in vivo 13C nuclear magnetic resonance. Environ Microbiol 9: 2694–2706.
Lopez-Vazquez CM, Hooijmans CM, Brdjanovic D, Gijzen HJ, van Loosdrecht MC . (2009). Temperature effects on glycogen accumulating organisms. Water Res 43: 2852–2864.
Liu W-T, Mino T, Nakamura K, Matsuo T . (1994). Role of glycogen in acetate uptake and polyhydroxyalkanoate synthesis in anaerobic-aerobic activated sludge with a minimized polyphosphate content. J Ferm Bioeng 77: 535–540.
Lu H, Oehmen A, Virdis B, Keller J, Yuan Z . (2006). Obtaining highly enriched cultures of Candidatus Accumulibacter phosphatis through alternating carbon sources. Water Res 40: 3838–3848.
McIlroy SJ, Kristiansen R, Albertsen M, Karst SM, Rossetti S, Nielsen JL et al (2013). Metabolic model for the filamentous ‘Candidatus Microthrix parvicella' based on genomic and metagenomic analyses. ISME J 7: 1161–1172.
McIlroy S, Seviour RJ . (2009). Elucidating further phylogenetic diversity among the Defluviicoccus-related glycogen-accumulating organisms in activated sludge. Environ Microbiol Rep 1: 563–568.
Mielczarek AT, Nguyen HT, Nielsen JL, Nielsen PH . (2013). Population dynamics of bacteria involved in enhanced biological phosphorus removal in Danish wastewater treatment plants. Water Res 47: 1529–1544.
Mino T, Van Loosdrecht MCM, Heijnen JJ . (1998). Microbiology and biochemistry of the enhanced biological phosphate removal process. Water Res 32: 3193–3207.
Nguyen HT, Le VQ, Hansen AA, Nielsen JL, Nielsen PH . (2011). High diversity and abundance of putative polyphosphate-accumulating Tetrasphaera-related bacteria in activated sludge systems. FEMS Microbiol Ecol 76: 256–267.
Nielsen AT, Liu W, Filipe C, Grady L Jr, Molin S, Stahl DA . (1999). Identification of a novel group of bacteria in sludge from a deteriorated biological phosphorus removal reactor. Appl Environ Microbiol 65: 1251–1258.
Nielsen PH, Mielczarek AT, Kragelund C, Nielsen JL, Saunders AM, Kong Y et al (2010). A conceptual ecosystem model of microbial communities in enhanced biological phosphorus removal plants. Water Res 44: 5070–5088.
Oehmen A, Carvalho G, Lopez-Vazquez CM, van Loosdrecht MC, Reis MA . (2010). Incorporating microbial ecology into the metabolic modelling of polyphosphate accumulating organisms and glycogen accumulating organisms. Water Res 44: 4992–5004.
Oehmen A, Yuan Z, Blackall LL, Keller J . (2005). Comparison of acetate and propionate uptake by polyphosphate accumulating organisms and glycogen accumulating organisms. Biotechnol Bioeng 91: 162–168.
Oehmen A, Lemos PC, Carvalho G, Yuan Z, Keller J, Blackall LL et al (2007). Advances in enhanced biological phosphorus removal: from micro to macro scale. Water Res 41: 2271–2300.
Oehmen A, Yuan Z, Blackall LL, Keller J . (2004). Short-term effects of carbon source on the competition of polyphosphate accumulating organisms and glycogen accumulating organisms. Water Sci Technol 50: (10): 139–144.
Oksanen et al (2011), Vegan: community ecology package. R package version 2.0-5.
Ostle A, Holt J . (1982). Nile blue A as a fluorescent stain for poly-beta-hydroxybutyrate. Appl Environ Microbiol 44: 238–241.
Portais JC, Delort AM . (2002). Carbohydrate cycling in micro-organisms: what can (13)C-NMR tell us? FEMS Microbiol Rev 26: 375–402.
Preiss J . (2006). Bacterial glycogen inclusions: enzymology and regulation of synthesis. In: Microbiology Monographs. Shively JM (ed). Springer-Verlag: Berlin, pp 71–108.
Rehm BH . (2003). Polyester synthases: natural catalysts for plastics. Biochem J 376: 15–33.
Rho M, Tang H, Ye Y . (2010). FragGeneScan: predicting genes in short and error-prone reads. Nucleic Acids Res 38: e191.
Richter M, Rosselló-Móra R . (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA 106: 19126–19131.
Saunders A, Mabbett A, McEwan A, Blackall L . (2007). Proton motive force generation from stored polymers for the uptake of acetate under anaerobic conditions. FEMS Microbiol Lett 274: 245–251.
Schroeder S, Petrovski S, Campbell B, McIlroy S, Seviour RJ . (2009). Phylogeny and in situ identification of a novel gammaproteobacterium in activated sludge. FEMS Microbiol Lett 297: 157–163.
Seviour RJ, McIlroy S . (2008). The microbiology of phosphorus removal in activated sludge processes- the current state of play. J Microbiol 46: 115–124.
Seviour RJ, Mino T, Onuki M . (2003). The microbiology of biological phosphorus removal in activated sludge systems. FEMS Microbiol Rev 27: 99–127.
Seviour T, Lambert LK, Pijuan M, Yuan Z . (2010). Structural determination of a key exopolysaccharide in mixed culture aerobic sludge granules using NMR spectroscopy. Environ Sci Technol 44: 8964–8970.
Seviour TW, Lambert LK, Pijuan M, Yuan Z . (2011). Selectively inducing the synthesis of a key structural exopolysaccharide in aerobic granules by enriching for Candidatus "Competibacter phosphatis". Appl Microbiol Biotechnol 92: 1297–1305.
Smolders GJF, van der Meij J, van Loosdrecht MCM, Heijnen JJ . (1994). Stoichiometric model of the aerobic metabolism of the biological phosphorus removal process. Biotechnol Bioeng 44: 837–848.
Vallenet D, Engelen S, Mornico D, Cruveiller S, Fleury L, Lajus A et al (2009). MicroScope: a platform for microbial genome annotation and comparative genomics. Database (Oxford) 2009: bap021.
Vallenet D, Labarre L, Rouy Z, Barbe V, Bocs S, Cruveiller S et al (2006). MaGe: a microbial genome annotation system supported by synteny results. Nucleic Acids Res 34: 53–65.
van Veen HW, Abee T, Kortstee GJ, Pereira H, Konings WN, Zehnder AJ . (1994). Generation of a proton motive force by the excretion of metal-phosphate in the polyphosphate-accumulating Acinetobacter johnsonii strain 210A. J Biol Chem 269: 29509–29514.
Wang X, Zeng RJ, Dai Y, Peng Y, Yuan Z . (2008). The denitrification capability of cluster 1 Defluviicoccus vanus-related glycogen-accumulating organisms. Biotechnol Bioeng 99: 1329–1336.
Wexler M, Richardson DJ, Bond PL . (2009). Radiolabelled proteomics to determine differential functioning of Accumulibacter during the anaerobic and aerobic phases of a bioreactor operating for enhanced biological phosphorus removal. Environ Microbiol 11: 3029–3044.
Wilmes P, Andersson AF, Lefsrud MG, Wexler M, Shah M, Zhang B et al (2008). Community proteogenomics highlights microbial strain-variant protein expression within activated sludge performing enhanced biological phosphorus removal. ISME J 2: 853–864.
Wolf A, Kramer R, Morbach S . (2003). Three pathways for trehalose metabolism in Corynebacterium glutamicum ATCC13032 and their significance in response to osmotic stress. Mol Microbiol 49: 1119–1134.
Wong M, Liu W . (2007). Ecophysiology of Defluviicoccus-related tetrad-forming organisms in an anaerobic-aerobic activated sludge process. Environ Microbiol 9: 1485–1496.
Wong M, Liu W . (2006). Microbial succession of glycogen accumulating organisms in an anaerobic-aerobic membrane bioreactor with no phosphorus removal. Water Sci Technol 54: (1): 29–37.
Wong M, Tan FM, Ng WJ, Liu W . (2004). Identification and occurrence of tetrad-forming Alphaproteobacteria in anaerobic-aerobic activated sludge processes. Microbiology 150: 3741–3748.
Wrighton KC, Thomas BC, Sharon I, Miller CS, Castelle CJ, VerBerkmoes NC et al (2012). Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science 337: 1661–1665.
Zeng RJ, Yuan Z, Keller J . (2003). Enrichment of denitrifying glycogen-accumulating organisms in anaerobic/anoxic activated sludge system. Biotechnol Bioeng 81: 397–404.
Zengin GE, Artan N, Orhon D, Chua AS, Satoh H, Mino T . (2010). Population dynamics in a sequencing batch reactor fed with glucose and operated for enhanced biological phosphorus removal. Bioresource Technol 101: 4000–4005.
Acknowledgements
The study was funded by Aalborg University, Krüger AS and the Danish Council for Strategic Research (EcoDesign-MBR). We thank JL Nielsen and M Stevenson for the MAR-FISH analysis. We also acknowledge the LABGeM team at Genoscope for providing access to their automated annotation pipeline as well as for database maintenance and technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
McIlroy, S., Albertsen, M., Andresen, E. et al. ‘Candidatus Competibacter’-lineage genomes retrieved from metagenomes reveal functional metabolic diversity. ISME J 8, 613–624 (2014). https://doi.org/10.1038/ismej.2013.162
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2013.162
Keywords
This article is cited by
-
Effect of Magnetic Field Enrichment Conditions on the Synthesis of Polyhydroxyalkanoates by Activated Sludge
Waste and Biomass Valorization (2024)
-
Biological and Chemical Approaches for Controlling Harmful Microcystis Blooms
Journal of Microbiology (2024)
-
Ethylmalonyl-CoA pathway involved in polyhydroxyvalerate synthesis in Candidatus Contendobacter
AMB Express (2022)
-
Development of microbial communities in biofilm and activated sludge in a hybrid reactor
Scientific Reports (2022)
-
Universal activity-based labeling method for ammonia- and alkane-oxidizing bacteria
The ISME Journal (2022)
