
Abstract
Coastal microbial mats are small-scale and largely closed ecosystems in which a plethora of different functional groups of microorganisms are responsible for the biogeochemical cycling of the elements. Coastal microbial mats play an important role in coastal protection and morphodynamics through stabilization of the sediments and by initiating the development of salt-marshes. Little is known about the bacterial and especially archaeal diversity and how it contributes to the ecological functioning of coastal microbial mats. Here, we analyzed three different types of coastal microbial mats that are located along a tidal gradient and can be characterized as marine (ST2), brackish (ST3) and freshwater (ST3) systems. The mats were sampled during three different seasons and subjected to massive parallel tag sequencing of the V6 region of the 16S rRNA genes of Bacteria and Archaea. Sequence analysis revealed that the mats are among the most diverse marine ecosystems studied so far and consist of several novel taxonomic levels ranging from classes to species. The diversity between the different mat types was far more pronounced than the changes between the different seasons at one location. The archaeal community for these mats have not been studied before and revealed a strong reaction on a short period of draught during summer resulting in a massive increase in halobacterial sequences, whereas the bacterial community was barely affected. We concluded that the community composition and the microbial diversity were intrinsic of the mat type and depend on the location along the tidal gradient indicating a relation with salinity.
Similar content being viewed by others

Introduction
Coastal microbial mats are nearly closed minimal ecosystems consisting of horizontal stratified groups or communities of microbes (Stal et al., 1985; van Gemerden, 1993). They are considered to be the modern analogs of fossil stromatolites, the oldest of which date back 3.5 billion years and therefore represent the oldest ecosystems known (Byerly et al., 1986). Coastal microbial mats are built by Cyanobacteria and are responsible for enriching the sediment with organic carbon. This process forms the basis for a complex microbial ecosystem in which a plethora of different functional groups of microorganisms accomplish a ‘joint venture’ (van Gemerden, 1993). The various metabolic capacities of this microbial community are responsible for the biogeochemical cycling of the elements. The sediment of microbial mats is also enriched with combined nitrogen through fixation of dinitrogen, an important property found so far in all studied microbial mats (Severin and Stal, 2010). In microbial mats, the capacity of photosynthetic carbon dioxide fixation often exceeds by far that of the fixation of dinitrogen (Stal et al., 2010), and this excess carbon is exuded as extracellular polymeric substances that form the matrix in which the microorganisms are embedded. Extracellular polymeric substances also serve as glue that produces a coherent structure that renders stability to the mat and the sediment surface (Grant and Gust, 1987). Microbial stabilization of the sediment and enrichment with organic carbon and nutrients form the basis for the colonization of salt marsh vegetation.
The North Sea beach of the Dutch barrier island Schiermonnikoog experienced spectacular changes over the past 20 years and transformed from a long stretched sandy beach into a broad salt marsh covering large areas of the island's North Sea coast (de Wit and van Gemerden, 1988). This ‘Green Beach’, as it is now coined, is an excellent example of coastal morphodynamics and protection. The bare and infertile sandy beaches are initially colonized by the oxygenic phototrophic Cyanobacteria, which form microbial mats and enrich the sediment with organic matter and nutrients (Stal et al. 1985). Subsequent colonization by vegetation further stabilizes the sediment and therewith increases its erosion threshold (Yallop et al., 1994). The accumulation of sediment by the salt marsh vegetation results in its elevation and increases the beach profile. Microbial mats of Schiermonnikoog have been studied using various techniques including microscopy (Stal et al., 1985; Visscher et al., 1991; van Gemerden, 1993; Dijkman et al., 2010), biogeochemical analysis involving microelectrode measurements (de Wit and van Gemerden, 1988; van Gemerden, 1993) and fatty acid profiling (Dijkman et al., 2010). Isolation efforts yielded cultures of several Cyanobacteria such as Microcoleus sp. and Lyngbya sp. and of Bacteria involved in the sulfur cycle such as Thiocapsa sp., Thiobacillus sp. and Desulfomicrobium sp. (Stal et al., 1985; de Wit and van Gemerden, 1988; Visscher et al., 1992; Wieringa et al., 2000). DNA sequence analysis of a 16S rRNA gene clone library revealed mainly Cyanobacteria, Proteobacteria and Bacteriodetes species (Severin et al., 2010). However, this library consisted of only ∼60 bacterial 16S rRNA gene sequences, leaving room for undersampling and statistical errors (Kemp and Aller, 2004) especially in this high-diversity microbial mat.
The aim of this study is to fully understand the diversity and biochemical complexity of these coastal microbial mats at a resolution that nearly captures the full diversity. The higher resolution will provide insight in the absence or presence/abundance of species present below the detection limit of previously used techniques and that are known to play a key role in biochemical processes (for example, anaerobic ammonium oxidation and methanotrophy). Moreover, little is known about the archaeal composition of these coastal microbial mats and may provide insight in their contribution to the microbial mats and especially the role of methanogenesis, a process strictly performed by Archaea (Thauer, 1998). We applied massive parallel 16S rRNA gene tag sequencing (Sogin et al., 2006) allowing the analysis of several thousands of small bacterial and archaeal 16S rRNA gene fragments, which tremendously increased the resolution of assessing microbial diversity.
Materials and methods
Sample site and sampling
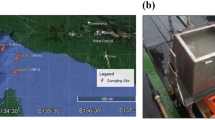
Samples were taken from microbial mats developing on the North Sea beaches of the Dutch barrier island Schiermonnikoog (Figure 1). The samples were taken during three different seasons (autumn 2007, spring 2008 and summer 2008) from three different microbial mat types located along the tidal gradient in close proximity of each other (Table 1). The stations are representatives of three different mat types that are each ∼100 m wide and cover the North Sea beach for over 5 km in length. Approximately 3–5 mm of the top layer of the sediment containing the whole microbial mat (∼20 cm2) was dissected, flash frozen in liquid nitrogen and stored at –80 °C.

Aerial picture and geographical location of the sampling stations (see insert) at the northwestern part of the North Sea coast of the Dutch barrier island Schiermonnikoog. Coordinates of the sampling points are presented in Table 1. See Materials and methods for a description of the stations. The pictures were obtained with Google Maps.
Nucleic acid extraction
Total community DNA was isolated from 1 g (wet weight) of mat using the UltraClean Soil DNA Isolation Kit (MoBio Laboratories, Inc., Carlsbad, CA, USA) according to the manufacturer's instructions for maximal yields. DNA yields were determined using spectrophotometry (Table 1) and the quality was checked by PCR amplification of the bacterial and archaeal 16S rRNA genes.
16S rRNA gene tag sequencing
Tag sequencing was performed as previously described (Sogin et al., 2006; Huber et al., 2007). In short, conserved sequences flanking the hypervariable V6 region of rRNAs served as primer sites to generate PCR amplicons. In order to capture the full diversity of bacterial 16S rRNA gene sequences, a cocktail of five primers was used at the 5′-end and four primers at the 3′-end of the V6 region (Table 2). For archaeal sequences, one 5′ primer and two 3′ primers were used (Table 2). Each PCR reaction was run in triplicate. The purified amplicon libraries were annealed to oligonucleotides that are complementary to an adaptor sequence that are tethered onto μm-size beads. The annealing conditions were set to favor one fragment per bead. PCR amplification of these fragments generating ∼10 million identical copies of a unique DNA template per bead. Subsequently, the beads carrying the single-stranded DNA clones were deposited into the wells of the PicoTiterPlate for pyrosequencing on a Roche Genome Sequencer (Roche Diagnostics GmbH/454 Life Sciences Corporation, Branford, CT, USA) (Sogin et al., 2006).
Sequence read analysis
Sequence reads of on average 60 bps were generated after trimming of the primer sequences from beginning and end of the raw data. Based on the assessment of pyrosequencing error rates, sequences of low quality were removed (Huse et al., 2007). Alignment and taxonomy assignment was performed using the tag mapping methodology GAST (global alignment for sequence taxonomy) (Sogin et al., 2006; Huse et al., 2007). Sequence reads were compared with a reference database of known 16S rRNA genes (obtained from SILVA and Ribosomal Database Project (RDP) databases) and taxonomically assigned according to the RDP classifier (Wang et al., 2007).
Statistical analysis
Taxonomic richness and diversity estimators were calculated using DOTUR/MOTHUR (www.mothur.org) software packages (Schloss and Handelsman, 2005; Schloss et al., 2009). We used the mean of the estimated richness for comparison between the samples. Cluster analysis of the community composition was performed using the statistical software package PAST (http://folk.uio.no/ohammer/past/) using a correlation matrix determined by the complement 1-r of Pearson's r correlation across the variables (Hammer et al., 2001).
Results
Description of the sampling sites
Station ST1 (Figure 1) is located in the supratidal area near the dunes and is mainly influenced by fresh water (rainfall). This station is inundated by sea water only occasionally at spring tide combined with northern winds. This area consist of small, young dunes, puddles of water and is densely vegetated with typical salt marsh plant species like Elymus farctus, Juncus gerardi, Glaux maritima, Ammophila arenaria and Scirpus maritimus. Microscopy revealed a high cyanobacterial diversity consisting of heterocystous (Nostoc, Calothrix and Anabaena) and non-heterocystous filamentous (Spirulina, Phormidium and Nodularia) Cyanobacteria as well as unicellular types (Synechocystis, Merismopedia and Gloeocapsa) (Severin and Stal, 2008; Dijkman et al., 2010). Diatoms were also common at this station. Station ST2 is situated near the low water mark, contains no vegetation and is continuously exposed to sea water due to tidal inundation. It receives only occasionally fresh water during rain showers at low tide. Here, mostly non-heterocystous Cyanobacteria, predominantly Lyngbya aestuarii and Leptolyngbya sp., were identified by microscopy, whereas diatoms were more abundant than at the other stations (Severin and Stal, 2008). Station ST3 is in between ST1 and ST2 and is characterized by microbial mats that receive fresh water and sea water (mostly at spring tides) at irregular intervals. ST3 forms the basis of a salt marsh ecosystem and is partly overgrown by only a few species of plants, mainly Salicornia sp. and Puccinellia distans. Microscopy revealed Microcoleus chthonoplastes as the dominant group of Cyanobacteria in ST3. To a lesser extent L. aestuarii and occasionally heterocystous and unicellular Cyanobacteria were found. The microbial mats at ST1 and ST3 contained a green layer of Cyanobacteria just below a thin layer of fine sand that had been recently deposited, whereas a green/brownish layer was observed at ST2. During summer and autumn, a distinct purple layer was noticed at ST1 and ST3 that was absent during spring and never noticed at ST2. At all stations, a black, iron sulfide-rich horizon was present at ∼5–10 mm depth.
General statistics
Using the bacterial V6 primers we obtained a total of 169 338 sequences, 92% of which were identified as Bacteria (Table 3). For each sample we obtained an average of 17 247 bacterial sequence reads. The archaeal V6 primers were less specific. On closer inspection, only 27 361 out of 85 415 reads (24%) were archaeal with an average of 3909 per sample. The majority of the reads appeared to belong to Eukarya. The frequency of bacterial singletons (unique reads that occurred only once in a sample divided by the total number of different sequence reads in that sample) was remarkably constant at 0.69±0.01. Archaeal reads revealed a similar conservation of the frequency of singletons, although at the higher value of 0.79±0.03.
Estimation of richness and diversity
Tags that differed by no more than 5% were clustered into operational taxonomic units (OTUs) to calculate rarefaction and nonparametric estimators (Table 4) (Schloss and Handelsman, 2005). The highest bacterial richness (the number of different OTUs in a sample) and diversity (Shannon's H index that takes into account the evenness of OTU distribution) were found during autumn and were lowest during spring with the exception of ST3 that had the lowest values during summer. For the archaeal fraction we only have a complete data set for station ST2 that showed a similar trend of highest richness and diversity in autumn and lowest in spring. In ST3, the archaeal richness was notably high in summer while having the lowest value for the diversity estimators.
Rarefaction analysis of the bacterial diversity in all stations at a 95% cutoff level revealed curves that did level of but did not yet reach an asymptote (Supplementary Figure S1), whereas nonparametric statistical analyses (Table 4) indicated that the observed richness covered between 60 and 66% of the estimated total bacterial richness. The rarefaction curves for archaeal richness also did not yet reach an asymptote (Supplementary Figure S2) and the observed richness covered 74–80% of the estimated archaeal richness. Overall, the observed bacterial richness was between 6- and 29-fold higher than the archaeal richness (Table 4). Shannon and Simpson diversity indices also indicated a higher diversity for the bacterial population relative to that of the archaeal population.
Spatial versus temporal diversity patterns in the bacterial community
Cluster analysis at the phylum level revealed a conservation of the community composition in time in a specific mat type. The stations ST3 and ST1 were more similar and clustered separately from station ST2 (Figure 2). The autumn and spring community compositions were more similar to each other than each of them to the summer community of ST2 and ST3, whereas for ST1 the spring and summer community compositions were more similar to each other than to the autumn community. Similar results were obtained when comparing the taxonomic composition at the order level (data not shown). A similar distribution was also found for the archaeal community; stations ST1 and ST3 clustered and their compositions were dissimilar from ST2.

Comparison of bacterial diversity between the different sampling points. (a) Cluster analysis of bacterial diversity at the phylum level using a correlation algorithm (the complement 1-r of Pearson's r correlation across the variables). (b) Stacked column graph representing the relative distribution of the dominant phyla in the different stations. The total number of sequence reads per sample is given left of the columns. (c) Stacked column graph representing the proteobacterial diversity. The double-headed arrow on top indicates the conditions with respect to salinity. Sample codes consist of the sampling station and the time point of sampling (Au, autumn; Sp, spring; Su, summer).
Taxonomic description
The obtained taxonomy data covered a broad spectrum of known bacterial phyla. Candidate phyla OP8, BRC1, OP11, OP3, OP10, TG1 and JS1 were represented by only a few sequences (<25). The dominant phyla in all samples belonged to Proteobacteria, Bacteriodetes, Cyanobacteria and Actinobacteria (Figure 2). A full description of the bacterial taxonomic data can be found in Supplementary Table S1. Table 5 lists the top 10 dominant groups per station, revealing large differences between stations ST1/ST3 and ST2, the latter being characterized by the absence of cyanobacterial sequences from this top 10 and by a more pronounced role for the Gammaproteobacteria. In contrast, Flavobacteriaceae, which were abundant in ST3 and ST2, did not appear in the top 10 of ST1. In addition to the dominant phyla, significant numbers of sequence reads related to Verrucomicrobia, Firmicutes, Planctomycetes, Chloroflexi, Gemmatimonadetes and Acidobacteria were found in all the three mat types. A small fraction (1.4–3.3%) of the reads could not be assigned below the domain level and were designated as Bacteria NA (not assigned). Among the Proteobacteria, the Alphaproteobacteria dominated ST1 (>70% of the Proteobacteria) and ST3 (>50% of the Proteobacteria; Figure 2). The Alphaproteobacteria consisted mainly of organisms belonging to the orders of Rhodobacterales and Sphingomonadales (Table 5). In station ST2 the α-(mainly Rhodobacterales), δ-(mainly Desulfobacterales and Myxococcales) and Gammaproteobacteria (mainly Chromatiales and Xanthomonadales) were more evenly distributed. A potential novel Gammaproteobacterium could not be assigned below the class level. Among the Betaproteobacteria, Burkholderiales dominated in ST1 and ST3 but were virtually absent in ST2. Only 49 reads affiliated to the Epsilonproteobacteria whereas 1624 reads could not be assigned below the Proteobacteria phylum level. Bacteriodetes were represented by Flavobacteriales and Sphingobacteriales. Sequences related to Flavobacteriales were particularly high in ST2, with a peak in the autumn population and a minimum during summer. A similar trend was observed in ST1 and ST3. The Sphingobacteriales peaked in the autumn populations in all three stations.
Cyanobacterial sequences were abundantly present in ST1 and ST3, and were dominated (71–96%) by members that we could not assign below the phylum level. Sequences belonging to the cyanobacterial groups GpIV, GpI, GXIII and Rivulariaceae were also well represented in ST1 and ST3. Other cyanobacterial sequences in these two stations were related to the Nostocales, Oscillatoriales, Prochlorales and Chroococcales. Station ST2 revealed only a few cyanobacterial sequences.
Among the Actinobacteria, Actinomycetales and NA actinobacterial order were dominating at all stations. Less abundant reads were assigned to the orders of Acidimicrobiales and Rubrobacterales. At the family level, the Actinomycetales were mainly represented by NA family with the highest number of reads in ST2, especially during autumn and summer. In contrast, the actinomycetal family of Microbacteriaceae were especially abundant in ST1 and ST3 and were virtually absent in ST2.
The archaeal diversity varied between the stations (Figure 3). A full description of the archaeal taxonomic data can be found in Supplementary Table S2. Station ST1 was dominated by Euryarchaeota (80–90%), mainly Methanobacteria (autumn population) or Methanomicrobia (summer population). The crenarchaeotal class Thermoprotei is only present in the autumn population of ST1. The summer population of ST1 contained an important fraction of NA Crenarchaeota and Euryarchaeota (13% and 10%, respectively). Station ST3 was dominated by Euryarchaeota of the class Halobacteria, especially in the summer where it accounted for 96% of the archaeal community. The spring community of ST3 contained Halobacteria, Methanomicrobia and a large fraction (29%) of NA Euryarchaeota. Station ST2 differs from the other two stations by having more NA Archaea, lower numbers of Euryarchaeota (42–56%) and more Crenarchaeota (11–39%). At the class level, the spring and summer communities of ST2 were dominated by Methanomicrobia, whereas the autumn community revealed more Thermoprotei. In ST2, Halobacteria were only present in low numbers.

Comparison of archaeal diversity between the different sampling points. (a) Cluster analysis of archaeal diversity at the class level using a correlation algorithm (the complement 1-r of Pearson's r correlation across the variables). (b) Stacked column graph representing the relative distribution of the dominant classes in the different stations. The total number of sequence reads per sample is given left of the columns. A distinction is made between the Crenarchaeota (shades of blue), the Euryarchaeota (purple to yellow) and the non-assigned (NA) Archaea (dark gray). The double-headed arrow on top indicates the extant conditions with respect to salinity. Sample codes consist of the sampling station and the time point of sampling (Au, autumn; Sp, spring; Su, summer).
Discussion
Richness analysis at a 95% cutoff value shows that it covered on average 62% (Bacteria) and 75% (Archaea) of the expected richness based on the Chao1 estimator (Table 4), and thus our data set gives a good representation of the actual microbial diversity. The average Shannon diversity index (H) calculated for the coastal mat samples indicates a relatively high diversity in comparison with eight different marine environments that were previously described by Sogin et al (2006). At a 0.05 OTU cutoff, the average H index was estimated to be 5.2±0.3 for the eight marine environments (Schloss et al., 2009), whereas our samples revealed an average H index of 5.9±0.4. This confirms previous reports that coastal microbial mats are among the most diverse marine ecosystems (Allen et al., 2009). The high diversity of microbial mats was attributed to the multitude of potential environmental niches and the large daily fluctuations of the key geochemical parameters (for example, pH, oxygen and sulfide) (Allen et al., 2009). Such conditions select for diverse microbial assemblages that on their turn drive diversity through niche creation, and hence forming a feedback between chemical complexity and biological diversity (Ley et al., 2006). The lower diversity in spring is in agreement with the succession of a young developing mat community that reaches maturity in summer and a climax during early autumn, after which the community becomes less productive, low in biomass and low in organic supply (Stal et al., 1985; Paerl et al., 1996). The autumn and winter mats are prone to destruction because of erosion by freezing, ice floes, storms or high sedimentation rates (Stal et al., 1985).
Cluster analysis of the bacterial composition at the level of phylum and order grouped ST1 and ST3 apart from ST2 (Figure 2). This reflects the difference between the marine, fully intertidal character of ST2 and the freshwater to brackish habitats of ST1 and ST3 that are located in supralittoral to high eulitoral, respectively, and irregularly inundated. This classification agrees with average salinities for these mat types that were previously determined by refractometry. During sampling campaigns held in spring, summer and autumn of 2003, the average salinity for the marine mat (ST2 type) was 57 psu (practical salinity units), the intermediate mat (ST3 type) was 45 psu and the freshwater mat (ST1 type) was 26 psu (LJ Stal, unpublished results). Especially the number of cyanobacterial sequences was strongly decreased at ST2 to the benefit of Bacteriodetes and Actinobacteria. A similar result was found in polar microbial mats where Proteobacteria rather than Cyanobacteria dominated the microbial population (Varin et al., 2010). The relative fractions of most other phyla remained unaltered. The archaeal composition also clustered per station but did not reveal the grouping of ST1 with ST3. Apparently, factors driving the bacterial community composition were different from those that affected the archaeal community. Seasonal variations were less pronounced in the bacterial communities and only slightly affected clustering. In both ST1 and ST3, the spring and summer bacterial populations were more similar to each other than to the autumn population. This is in agreement with general observations of seasonality of annual microbial mat communities.
The archaeal community composition in ST2 appeared to be stable, whereas the composition in ST1 and ST3 underwent large fluctuations. In ST1 the Methanobacteria dominated the community in autumn and was replaced in the following summer by Methanomicrobia. Long-term analysis over several consecutive years should indicate whether this was part of a yearly shift in methanogenic communities in response to mat maturation or whether this was a one-time event. For the ecosystem function this change has probably little impact as both are involved in methanogenesis. In ST3, the archaeal fraction was dominated by Halobacteria. Notably in summer the observed richness was for 96% attributed to halophilic Archaea. These summer samples were taken after some weeks of high temperatures and low rainfall causing desiccation and increased local salinities, giving way for halophilic organisms. Sequences of a typical bacterial halophilic genus, Halobacillus, were slightly increased in the summer community of ST3, whereas the halotolerant Halochromatium was mainly found in the summer community of ST1. Salinibacter, although present in ST3, did not reveal higher numbers in the summer community. Despite the harsher conditions during summer that favor growth of halophiles, the diversity in ST3 was only slightly lower (H=5.8) than in autumn and spring (H=6.1). Possibly, the non-halotolerant microorganisms were protected from desiccation by the copious amounts of extracellular polymeric substances. Alternatively, the halophilic Archaea may have been confined to hypersaline micro-habitats or salt crystals within the extracellular polymeric substance matrix formed during exsiccation (Norton and Grant, 1988).
Taxonomic inventory
The dominant phyla for all stations consisted of the Proteobacteria, Bacteroidetes, Cyanobacteria and Actinobacteria and are known to be abundant in marine ecosystems including microbial mats (Green et al., 2008; Heyl et al., 2010; Severin et al., 2010). However, Actinobacteria were previously not detected in the Schiermonnikoog mats (Severin et al., 2010). This may have been because of the primers that were less suitable for amplification of the 16S rRNA gene of this group. A small fraction (1.4–3.3%) of the sequence reads could not be affiliated with any of the known bacterial phyla and may be obtained from representatives of novel, not yet described bacterial phyla. Each coastal mat type also contained considerable numbers of sequence reads related to three candidate divisions (TM7, WS3 and OD1) of which no cultivated representatives exist to date. Hitherto, no distinct ecological function has been assigned to members of the candidate division TM7 that are found in a wide range of environments, including terrestrial (soils, rhizosphere and peat bog) and aquatic (groundwater, fresh water, sea water, and deep-sea sediments) habitats (Rheims et al., 1996; Hugenholtz et al., 2001). Members of the candidate division WS3 members have been found associated with methanogenic environments (Dojka et al., 1998). The OD1 division is widespread in environments dominated by sulfur, such as marine environments and sulfureta (Harris et al., 2004). At the tail of bacterial diversity comprising <70 reads in the 9 samples, we found members of the Nitrospira, Fibrobacteres, Fusobacteria, Tenericutes, Deferribacteres and of several other candidate divisions (WS3, TG1, OP3-8-10-11, BRC1, TM6) originally detected in extreme, low-diversity environments (Ohkuma and Kudo, 1996; Rheims et al., 1996; Dojka et al., 1998; Hugenholtz et al., 1998; Derakshani et al., 2001).
Functional diversity
Functional diversity was derived from those sequence reads that were assigned to a family, genus or higher taxonomic levels of which the functional properties are strongly conserved. For example, sequence reads assigned as Rivularia sp. are considered to be derived from dinitrogen-fixing photoautotrophes (Livingstone et al., 1984). Obviously, this might underestimate the actual number of contributors to ecosystem functions because potential functional groups may be hidden in sequences that could not be assigned beyond the taxonomic level of phyla, class or order. Bacterial abundance grouped according to their function (Figure 4) revealed that approximately half were chemoorganotrophs that presumably grow at the expense of the abundantly present carbohydrates excreted by the primary producers Cyanobacteria and diatoms. More than 30% of the bacterial community consisted of photoautotrophs (Cyanobacteria) and photoheterotrophs (mainly Rhodobacterales). Sulfate-reducing bacteria (SRB) and sulfide-oxidizing bacteria varied from 1.4% to 5% of the total bacterial sequences, and this relatively low number surprised us, considering the fact that the sulfur cycle is so prominently present in marine microbial mats.

Distribution of bacterial functional groups at the different sampling points. Functional characteristics were only assigned to those bacterial orders for which a conserved function is well documented. Methanol, methanol-degrading bacteria; SOB, sulfide-oxidizing bacteria.
Because we used the cyanobacterial identification following the RDP classifier (Wang et al., 2007), which is based on the classification described in Bergey's Manual of Systematic Bacteriology (Castenholz, 2001), the taxonomic breakdown of the Cyanobacteria is complex. Unfortunately, many Cyanobacteria in this classification scheme are not described beyond the family level and are distributed in 13 different families and genera (GpI–GpXIII). Furthermore, cyanobacterial taxonomy was historically based on morphological features according to the Botanical Code, whereas 16S rRNA sequence-based taxonomy is hampered by incorrect naming (Komarek, 2010). Sequences related to several groups and families of Cyanobacteria (GpI, GpXIII, Rivularia, Microcoleus, Chroococcales, and GpX) dominated in ST3, whereas they occurred in much lower numbers in the other stations. At this moment, we can only speculate on the cause for this observation. Possible explanations are variations in predation stress in the different stations, preference of the listed Cyanobacteria for a brackish environment rather than marine or freshwater or a different response to desiccation stress. Finding the actual cause will require dedicated experimentation. Microcoleus sp., the microscopically dominant species in ST3 and an important contributor to mat stabilization and perhaps N2 fixation (Bolhuis et al., 2010), was confined to the autumn community but did not appear abundantly in the 16S rRNA gene tag analysis. Also, Leptolyngbya sp. was a dominant group of Cyanobacteria as observed by microscopy in ST2 (Severin and Stal, 2008), but this genus also did not come up abundantly in our library. The Cyanobacteria may have been out-competed by diatoms that were present in large numbers in ST2. Typical marine Cyanobacteria only found in ST2 belong to GpVII (Halothece and Euhalothece) (Cyanobacteria normally found in hypersaline environments) and to GpVIII (Myxosarcina and Pleurocapsa).
In addition to Cyanobacteria, many other diazotrophic bacteria are present and may play an important role in N2 fixation (Severin et al., 2010). Apart from the Cyanobacteria, most diazotrophic species were found in ST1 and ST3, of which Rhizobiales, Burkholderiales and Rhodobacterales are known to be associated with plant roots. The intertidal station ST2, which is devoid of plants, lacks those species. A well-known cosmopolitan, plant root-associated N2-fixing genus Frankia, common in many soil types (Normand and Fernandez, 2009), was not found. Various Archaea like Methanosarcina were found in all stations and may also contribute to N2 fixation (Belay et al., 1984).
Whether anaerobic ammonium oxidation (anammox) (Mulder et al., 1995) takes place in these mats remains uncertain as no sequence reads were found directly related to the known anammox species among the Planctomycetes. However, the largest fraction of Planctomycetes (∼2500 reads) could not be assigned below the family level and at this point we cannot exclude that it may contain a hitherto unknown genus involved in the anammox process.
Anoxygenic phototrophic bacteria occur below the cyanobacterial layer. Overall, the Chloroflexi were mainly found in the freshwater stations ST1 and ST3, although a yet unidentified class within the Chloroflexi was most abundant in the marine mat of ST2. Furthermore, a large fraction (>70%) of the Chloroflexi belonged to the non-photosynthetic families of Anaerolineae and Caldilineae (Sekiguchi et al., 2003). OTUs related to Chloronema were only found in ST1, although at low numbers (<0.25%). Alphaproteobacterial purple non-sulfur bacteria (PNSB) and the gammaproteobacterial purple sulfur bacteria (PSB; Chromatiales) were found in all stations. Among the PNSB, Rhodobacter was the dominant genus in all three stations but preferentially in ST1. Other PNSB (for example, Rhodobaca and several Rhodospirillaceae) were present at lower numbers and also preferred the low-salinity microbial mats. Two previously characterized marine PSB, Marichromatium (Imhoff et al., 1998) and Ectothiorhodospiraceae (Imhoff, 2006) were indeed mainly found in ST2 while the halophilic Halochromatium peaked in the summer community of ST1, probably as a result of the extended time of drought before sampling. The typical freshwater genera (Allochromatium, Thiocapsa) were accordingly virtually absent in ST2. Sequences of the PSB Thiocapsa roseopersicina and the colorless chemolithoautotrophic sulfur bacterium Thiobacillus thiooxidans were found in relatively small numbers, which is in contrast to previous studies where these were found to be the dominant sulfur bacteria in microbial mats (de Wit and van Gemerden, 1988; van Gemerden et al., 1989; Visscher et al., 1992). However, these authors used most probable number techniques to enumerate these organisms, and it is possible that other types of organisms carry out the metabolism that was attributed to Thiocapsa sp. and Thiobacillus sp. Neither Thiocapsa sp. nor Thiobacillus sp. were found in 16S rRNA gene clone libraries from the same site (Severin et al., 2010), confirming our observations. The PSB showed a climax community during summer and their numbers were low during spring, which is in agreement with the absence of a visible purple layer in the spring samples.
Aerobic anoxygenic phototrophic bacteria, all belonging to the Alphaproteobacteria, were also found. These metabolically versatile heterotrophs cannot use light as the sole source of energy and therefore cannot grow autotrophically and are incapable of growing photosynthetically under anaerobic conditions (Yurkov and Beatty, 1998). The previously identified marine Erythrobacter and Roseobacter were found at all three stations, suggesting the presence of strains more tolerant to lower salinities. The freshwater Porphyrobacter and Sandaracinobacter were, as expected, confined to ST1 and ST3.
In contrast to the PSB, SRB are present throughout the mat and may even be found in the aerobic cyanobacterial layer (Krekeler et al., 1998). SRB use low-molecular organic compounds as substrates that are excreted by the Cyanobacteria through fermentation and photorespiration (Stal, 1995). SRB produce sulfide as the result of the dissimilatory reduction of sulfate that may precipitate as iron-sulfide or pyrite. This renders the typical black horizons in the deeper permanent anoxic layers of the mat (van Gemerden, 1993). The most important group of SRB is related to Desulfomicrobiaceae, members of which have been isolated from nearby locations (Wieringa et al., 2000). We did not found sequences belonging to this group in ST2, which might suggest a preference for low-salinity environments. Nevertheless, ST2 harbored the highest total number of SRB, especially Desulfobulbaceae, Desulfobacteraceae and Desulfuromonadaceae. Desulfosarcina, which is known to inhabit brackish to marine sediments (Kuever et al., 2005), and NA Desulfobulbaceae were confined to the marine station. Desulfonema, Desulfomicrobium and Desulfobacter, which are known to thrive in both freshwater and marine sediments, were virtually absent from ST2.
The Actinobacteria represented a large fraction of the microbial diversity in our mats. Increasingly more marine Actinobacteria are being discovered (Bull and Stach, 2007) and as shown here are also abundant in microbial mats. In contrast to several other phyla, the numbers of sequence reads related to Actinobacteria were more equally distributed over the three stations with the highest content in ST2 (14%) and lowest in ST3 (9%). The most abundant group of Actinobacteria could not be assigned beyond the class level followed by NA Actinomycetales and Microbacteriaceae. This suggests that novel species are present in our mats. The ability of Actinobacteria to produce a wide variety of extracellular hydrolytic enzymes, responsible for the breakdown of complex organic matter, makes them central organisms in the decomposition of carbon, notably in the degradation of chitin, the second most abundant organic substance in nature and especially in marine ecosystems produced by Crustacea (LeCleir et al., 2004). This functional group has received little attention in microbial mats, notwithstanding their importance.
Methanogenesis and methanotrophy are processes occurring in a wide variety of ecosystems. In sulfate-rich marine sediments, methanogens are readily out-competed by the SRB when competing for substrates such as acetate and hydrogen (Oremland et al., 1982). However, methanogens can use noncompetitive substrates such as methylamines, dimethylsulfoniopropionate and dimethylsulfide that are poorly used by SRB and are present in high concentrations in microbial mats (Oremland et al., 1982; Visscher et al., 1991; Jonkers et al., 1998). Methanogens all belong to the Archaea and considerable numbers of sequence reads related to methanogen sequences were present in all three stations, suggesting that methanogenesis is important in our mats. Methanogens related to Methanocorpusculum, Methanospirillum and Methanobacterium were restricted to ST1, whereas Methanomicrobiaceae were mostly found in station ST2. In ST1, Methanobacteria dominated the autumn community but were replaced by Methanomicrobia during the following summer. This change in gene pool would presumably not have affected ecosystem function with respect to methanogenesis. Sequence reads assigned as originated from methanotrophic bacteria were present at very low numbers and presumably did not play an important role in methane consumption in the investigated microbial mats. Methane may rather be emitted from the mats into the air or may be oxidized anaerobically by methanogenic Archaea (for example, Methanosarcinales) via reversed methanogenesis in a tight consortium with SRB (Boetius et al., 2000).
In summary, stabilization and fertilization of the bare sand by the dinitrogen-fixing microbial mats provides the prerequisites for vegetation to colonize sandy beaches. Not only did we confirm the major functional groups and geochemical processes in microbial mats that have been established before (Stal et al., 1985; van Gemerden, 1993; Severin et al., 2010), we also unveiled a previously unpredicted richness and diversity of Bacteria and Archaea. Among the marine ecosystems studied by massive parallel 16S rRNA gene tag sequencing, the microbial mats are the most diverse. Previous studies suggested species richness in the order of ∼50 OTUs at 97% identity and ∼500 OTUs at 100% identity (Severin et al., 2010). Here we have found a more realistic estimator for bacterial richness of ∼3000 OTUs at 95% and ∼12 000 OTUs at 100% identity. At 100% identity, this would largely describe ‘within-species diversity’ resulting in ecotypes of species adapted to their particular microniches. This study also suggests that we have uncovered the tip of the iceberg with respect to biochemical potential and the possible complex reactions between the different members of Bacteria and Archaea of the mat, while a yet unknown micro-eukaryotic and viral diversity still needs to be explored.
References
Allen MA, Goh F, Burns BP, Neilan BA . (2009). Bacterial, archaeal and eukaryotic diversity of smooth and pustular microbial mat communities in the hypersaline lagoon of Shark Bay. Geobiology 7: 82–96.
Belay N, Sparling R, Daniels L . (1984). Dinitrogen fixation by a thermophilic methanogenic bacterium. Nature 312: 286–288.
Boetius A, Ravenschlag K, Schubert CJ, Rickert D, Widdel F, Gieseke A et al. (2000). A marine microbial consortium apparently mediating anaerobic oxidation of methane. Nature 407: 623–626.
Bolhuis H, Severin I, Confurius-Guns V, Wollenzien UI, Stal LJ . (2010). Horizontal transfer of the nitrogen fixation gene cluster in the cyanobacterium Microcoleus chthonoplastes. ISMEJ 4: 121–130.
Bull AT, Stach JEM . (2007). Marine Actinobacteria: new opportunities for natural product search and discovery. Trends Microbiol 15: 491–499.
Byerly GR, Lower DR, Walsh MM . (1986). Stromatolites from the 3,300-3,500-Myr Swaziland Supergroup, Barberton Mountain Land, South Africa. Nature 319: 489–491.
Castenholz RW (ed). (2001). Phylum BX. Cyanobacteria. Oxygenic Photosynthetic Bacteria. Springer-Verlag: New York.
de Wit R, van Gemerden H . (1988). Interactions between phototrophic bacteria in sediment ecosystems. Aquat Ecol 22: 135–145.
Derakshani M, Lukow T, Liesack W . (2001). Novel bacterial lineages at the (sub)division level as detected by signature nucleotide-targeted recovery of 16S rRNA genes from bulk soil and rice roots of flooded rice microcosms. Appl Environ Microbiol 67: 623–631.
Dijkman NA, Boschker HTS, Stal LJ, Kromkamp JC . (2010). Composition and heterogeneity of the microbial community in a coastal microbial mat as revealed by the analysis of pigments and phospholipid-derived fatty acids. J Sea Res 63: 62–70.
Dojka MA, Hugenholtz P, Haack SK, Pace NR . (1998). Microbial diversity in a hydrocarbon- and chlorinated-solvent-contaminated aquifer undergoing intrinsic bioremediation. Appl Environ Microbiol 64: 3869–3877.
Grant J, Gust G . (1987). Prediction of coastal sediment stability from photopigment content of mats of purple sulfur bacteria. Nature 330: 244–246.
Green SJ, Blackford C, Bucki P, Jahnke LL, Prufert-Bebout L . (2008). A salinity and sulfate manipulation of hypersaline microbial mats reveals stasis in the cyanobacterial community structure. ISMEJ 2: 457–470.
Hammer, Harper DAT, Ryan PD . (2001). Paleontological statistics software package for education and data analysis. Palaeontol Electron 4: 9.
Harris JK, Kelley ST, Pace NR . (2004). New perspective on uncultured bacterial phylogenetic division OP11. Appl Environ Microbiol 70: 845–849.
Heyl K, Woelfel J, Schumann R, Karsten U . (2010). Microbial mats from wind flats of the Southern Baltic Sea. In: Seckbach J, Oren A (eds). Microbial Mats. Springer: The Netherlands, pp 301–319.
Huber JA, Mark Welch DB, Morrison HG, Huse SM, Neal PR, Butterfield DA et al. (2007). Microbial population structures in the deep marine biosphere. Science 318: 97–100.
Hugenholtz P, Pitulle C, Hershberger KL, Pace NR . (1998). Novel division level bacterial diversity in a Yellowstone Hot Spring. J Bacteriol 180: 366–376.
Hugenholtz P, Tyson GW, Webb RI, Wagner AM, Blackall LL . (2001). Investigation of candidate division TM7, a recently recognized major lineage of the domain Bacteria with no known pure-culture representatives. Appl Environ Microbiol 67: 411–419.
Huse SM, Huber JA, Morrison HG, Sogin ML, Welch DM . (2007). Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol 8: R143.
Imhoff JF . (2006). The family Ectothiorhodospiraceae. In: Dworkin M, Falkow S, Rosenberg E, Schleifer KH, Stackebrandt E (eds). The Prokaryotes: A Handbook on the Biology of Bacteria, 3rd edn, vol. 6. Springer: New York, pp 874–886.
Imhoff JF, Suling J, Petri R . (1998). Phylogenetic relationships among the Chromatiaceae, their taxonomic reclassification and description of the new genera Allochromatium, Halochromatium, Isochromatium, Marichromatium, Thiococcus, Thiohalocapsa and Thermochromatium. Int J Syst Bacteriol 48: 1129–1143.
Jonkers HM, Koopmans GF, van Gemerden H . (1998). Dynamics of dimethyl sulfide in a marine microbial mat. Microb Ecol 36: 93–100.
Kemp P, Aller J . (2004). Estimating prokaryotic diversity: when are 16S rDNA libraries large enough? Limnol Oceanogr Methods 2: 114–125.
Komarek J . (2010). Recent changes (2008) in Cyanobacteria taxonomy based on a combination of molecular background with phenotype and ecological consequences (genus and species concept). Hydrobiologia 639: 245–259.
Krekeler D, Teske A, Cypionka H . (1998). Strategies of sulfate-reducing bacteria to escape oxygen stress in a cyanobacterial mat. FEMS Microbiol Ecol 25: 89–96.
Kuever J, Rainey FA, Widdel F . (2005). Genus X. Desulfosarcina Widdel 1981, 382VP(Effective publication: Widdel 1980, 382). In: Brenner DJ, Krieg NR, Staley JT, Garrity GM (eds) Bergey's Manual of Systematic Bacteriology 2nd edn. Springer: New York, pp 988–992.
LeCleir GR, Buchan A, Hollibaugh JT . (2004). Chitinase gene sequences retrieved from diverse aquatic habitats reveal environment-specific distributions. Appl Environ Microbiol 70: 6977–6983.
Ley RE, Harris JK, Wilcox J, Spear JR, Miller SR, Bebout BM et al. (2006). Unexpected diversity and complexity of the Guerrero Negro hypersaline microbial mat. Appl Environ Microbiol 72: 3685–3695.
Livingstone D, Pentecost A, Whitton BA . (1984). Diel variations in nitrogen and carbon dioxide fixation by the blue-green alga Rivularia in an upland stream. Phycologia 23: 125–133.
Mulder A, van de Graaf AA, Robertson LA, Kuenen JG . (1995). Anaerobic ammonium oxidation discovered in a denitrifying fluidized bed reactor. FEMS Microbiol Ecol 16: 177–183.
Normand P, Fernandez M . (2009). Evolution and diversity of Frankia. In: Pawlowski K (ed.). Prokaryotic Symbionts in Plants, vol 8. Springer: Berlin/Heidelberg, pp 103–125.
Norton CF, Grant WD . (1988). Survival of Halobacteria within fluid inclusions in salt crystals. J Gen Microbiol 134: 1365–1373.
Ohkuma M, Kudo T . (1996). Phylogenetic diversity of the intestinal bacterial community in the termite Reticulitermes speratus. Appl Environ Microbiol 62: 461–468.
Oremland RS, Marsh LM, Polcin S . (1982). Methane production and simultaneous sulphate reduction in anoxic, salt marsh sediments. Nature 296: 143–145.
Paerl HW, Fitzpatrick M, Bebout BM . (1996). Seasonal nitrogen fixation dynamics in a marine microbial mat: potential roles of Cyanobacteria and Microheterotrophs. Limnol Oceanogr 41: 419–427.
Rheims H, Rainey FA, Stackebrandt E . (1996). A molecular approach to search for diversity among bacteria in the environment. J Ind Microbiol Biotechnol 17: 159–169.
Schloss PD, Handelsman J . (2005). Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol 71: 1501–1506.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537–7541.
Sekiguchi Y, Yamada T, Hanada S, Ohashi A, Harada H, Kamagata Y . (2003). Anaerolinea thermophila gen. nov., sp. nov. and Caldilinea aerophila gen. nov., sp. nov., novel filamentous thermophiles that represent a previously uncultured lineage of the domain Bacteria at the subphylum level. Int J Syst Evol Microbiol 53: 1843–1851.
Severin I, Acinas SG, Stal LJ . (2010). Diversity of nitrogen-fixing bacteria in cyanobacterial mats. FEMS Microbiol Ecol 73: 514–525.
Severin I, Stal LJ . (2008). Light dependency of nitrogen fixation in a coastal cyanobacterial mat. ISMEJ 2: 1077–1088.
Severin I, Stal LJ . (2010). Diazotrophic microbial mats. In: Seckbach J, Oren A (eds). Microbial Mats. Springer: The Netherlands, pp 321–339.
Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR et al. (2006). Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc Natl Acad Sci USA 103: 12115–12120.
Stal LJ . (1995). Physiological ecology of Cyanobacteria in microbial mats and other communities. New Phytol 131: 1–32.
Stal LJ, Gemerden H, Krumbein WE . (1985). Structure and development of a benthic marine microbial mat. FEMS Microbiol Lett 31: 111–125.
Stal LJ, Severin I, Bolhuis H . (2010). The ecology of nitrogen fixation in cyanobacterial mats. Adv Exp Med Biol 675: 31–45.
Thauer RK . (1998). Biochemistry of methanogenesis: a tribute to Marjory Stephenson. 1998 Marjory Stephenson Prize Lecture. Microbiology 144: 2377–2406.
van Gemerden H . (1993). Microbial mats: a joint venture. Mar Geol 113: 3–25.
van Gemerden H, de Wit R, Tughan CS, Herbert RA . (1989). Development of mass blooms of Thiocapsa roseopersicina on sheltered beaches on the Orkney Islands. FEMS Microbiol Lett 62: 111–118.
Varin T, Lovejoy C, Jungblut AD, Vincent WF, Corbeil J . (2010). Metagenomic profiling of Arctic microbial mat communities as nutrient scavenging and recycling systems. Limnol Oceanogr 55: 1901–1911.
Visscher PT, Quist P, van Gemerden H . (1991). Methylated sulfur compounds in microbial mats: in situ concentrations and metabolism by a colorless sulfur bacterium. Appl Environ Microbiol 57: 1758–1763.
Visscher PT, van den Ende FP, Schaub BEM, van Gemerden H . (1992). Competition between anoxygenic phototrophic bacteria and colorless sulfur bacteria in a microbial mat. FEMS Microbiol Lett 101: 51–58.
Wang Q, Garrity GM, Tiedje JM, Cole JR . (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73: 5261–5267.
Wieringa EBA, Overmann J, Cypionka H . (2000). Detection of abundant sulphate-reducing bacteria in marine oxic sediment layers by a combined cultivation and molecular approach. Environ Microbiol 2: 417–427.
Yallop ML, de Winder B, Paterson DM, Stal LJ . (1994). Comparative structure, primary production and biogenic stabilization of cohesive and non-cohesive marine sediments inhabited by microphytobenthos. Estuar Coast Shelf Sci 39: 565–582.
Yurkov VV, Beatty JT . (1998). Aerobic anoxygenic phototrophic bacteria. Microbiol Mol Biol Rev 62: 695–724.
Acknowledgements
We thank the International Census of Marine Microbes for funding the pyrosequencing and data analysis. We especially thank Dr Mitchell L Sogin and Dr Linda Amaral Zettler of the Marine Biological Laboratory at Woods Hole (MA, USA) for their support in pyrosequencing and initial data analysis. This is publication 5031 of the NIOO-KNAW.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website
Rights and permissions
About this article
Cite this article
Bolhuis, H., Stal, L. Analysis of bacterial and archaeal diversity in coastal microbial mats using massive parallel 16S rRNA gene tag sequencing. ISME J 5, 1701–1712 (2011). https://doi.org/10.1038/ismej.2011.52
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2011.52
Keywords
This article is cited by
-
Bacterial community network complexity and role of stochasticity decrease during primary succession
Soil Ecology Letters (2024)
-
Impact of meltwater flow intensity on the spatiotemporal heterogeneity of microbial mats in the McMurdo Dry Valleys, Antarctica
ISME Communications (2023)
-
Diversity pattern of marine culturable heterotrophic bacteria in a region with coexisting upwelling and mud banks in the southeastern Arabian Sea
Environmental Science and Pollution Research (2022)
-
Transcriptomic evidence for versatile metabolic activities of mercury cycling microorganisms in brackish microbial mats
npj Biofilms and Microbiomes (2021)
-
Prokaryotic diversity of tropical coastal sand dunes ecosystem using metagenomics
3 Biotech (2021)
