
Abstract
Quorum-sensing signal autoinducer 2 (AI-2) stimulates Escherichia coli biofilm formation through the motility regulator MqsR that induces expression of the putative transcription factor encoded by yncC. Here, we show that YncC increases biofilm formation by repressing overproduction of the exopolysaccharide identified as colanic acid (corroborated by decreasing mucoidy and increased sensitivity to bacteriophage P1 infection). Differential gene expression and gel shift assays demonstrated that YncC is a repressor of the predicted periplasmic protein-encoding gene, ybiM, which was corroborated by the isogenic yncC ybiM double mutation that repressed the yncC phenotypes (biofilm formation, colanic acid overproduction, mucoidy and bacteriophage resistance). Through nickel-enrichment DNA microarrays and additional gel shift assays, we found that the putative transcription factor B3023 (directly upstream of mqsR) binds the yncC promoter. Overexpressing MqsR, AI-2 import regulators LsrR/LsrK and AI-2 exporter TqsA induced yncC transcription, whereas the AI-2 synthase LuxS and B3023 repressed yncC. MqsR has a toxic effect on E. coli bacterial growth, which is partially reduced by the b3023 mutation. Therefore, AI-2 quorum-sensing control of biofilm formation is mediated through regulator MqsR that induces expression of the transcription factor YncC. YncC inhibits the expression of periplasmic YbiM, which prevents overproduction of colanic acid (excess colanic acid causes mucoidy) and prevents YbiM from inhibiting biofilm formation.
Similar content being viewed by others

Introduction
Escherichia coli biofilm development is a complex process with at least five developmental stages; initial reversible attachment of planktonic cells to a solid surface, transition from reversible to irreversible attachment, early development of biofilm architecture, development of microcolonies into mature biofilm and dispersion of cells from the biofilm to return to the planktonic state (Van Houdt and Michiels, 2005). Hundreds of genes are differentially expressed during this biofilm development process (Schembri et al., 2003; Beloin et al., 2004; Ren et al., 2004a) and bacterial signaling such as quorum and metabolite sensing and catabolite repression are involved in this process (Jackson et al., 2002; Stanley and Lazazzera, 2004; Camilli and Bassler, 2006).
One recently-characterized metabolite biofilm signal is indole, which represses biofilm formation of E. coli by repressing motility, inducing the sensor of the quorum-sensing signal autoinducer-1 and influencing acid resistance (Lee et al., 2007b). A series of genes including bssR/bssS, bhsA and ariR have been identified as regulating biofilm formation through indole (Domka et al., 2006; Lee et al., 2007c; Zhang et al., 2007), and hydroxy indoles have been shown to be signals for pathogenic E. coli (Bansal et al., 2007; Lee et al., 2007a).
Another signal controlling the biofilm development of E. coli is autoinducer 2 (AI-2) (González Barrios et al., 2006), a bacterial species-nonspecific signal used by both Gram-negative and Gram-positive bacteria and synthesized by S-ribosylhomocysteine lyase (LuxS) (Schauder et al., 2001). LuxS converts S-ribosyl-homocysteine into homocysteine and 4,5-dihydroxy-2,3-pentanedione (DPD), which cyclizes spontaneously into a family of AI-2 molecules (Lombardia et al., 2006). As a bacterial communication signal, AI-2 is exported by TqsA (Herzberg et al., 2006) and internalized by a lsr operon-encoded system (Taga et al., 2003), and then controls a variety of genes (DeLisa et al., 2001b; Xavier and Bassler, 2003; Ren et al., 2004b). The lsr operon of seven genes lsrACDBFGE is induced by phospho-AI-2 and regulated by LsrR, LsrK and GlpDK (Taga et al., 2001, 2003; Xavier and Bassler, 2005). The regulator LsrR represses the AI-2 uptake operon lsr, which is derepressed by the binding of phospho-AI-2 to LsrR (Taga et al., 2003). Another regulator, LsrK, a cytoplasmic kinase, phosphorylates internal AI-2 into an activated molecule (Xavier and Bassler, 2005). The glycerol uptake and metabolism system encoded by glpDFK also influences AI-2 signaling by regulating lsr transcription through LsrR (Xavier and Bassler, 2005).
Some insights have been gained as to how AI-2 controls biofilm formation. In E. coli, AI-2 stimulates biofilm formation and changes its architecture by stimulating flagellar motility via the quorum-sensing regulator MqsR that acts through the two-component motility regulatory system QseBC (González Barrios et al., 2006), which transcriptionally regulates FlhDC, the master regulator of flagella and motility genes fliLMNOPQR, fliAZ, flhBA and flgABCDMN (Liu and Matsumura, 1994; Claret and Hughes, 2002; Clarke and Sperandio, 2005). This result is consistent with the recent finding that in the oral bacterium Aggregatibacter actinomycetemcomitans, AI-2 regulates its biofilm formation most likely through its QseBC system (Shao et al., 2007). Also, in the human gastric pathogen Helicobacter pylori, AI-2 controls motility by controlling genes upstream of the motility and flagellar regulator FlhA (Rader et al., 2007). Further proof that AI-2 controls motility in different genera is that AI-2 regulates transcription of the flagellin gene, flaA, in the human pathogen Campylobacter jejuni (Jeon et al., 2003). In addition, BssR/BssS regulate E. coli biofilms by influencing AI-2 and indole concentrations in a divergent manner (Domka et al., 2006), and AI-2 regulates the dual-species biofilm formation of two Gram-positive human oral commensal bacterial strains, Actinomyces naeslundii T14V and Streptococcus oralis 34, at an optimum concentration (Rickard et al., 2006).
Details of how MqsR functions are still not clear. MqsR was first shown to be highly induced in biofilms by Ren et al. (2004a). MqsR has also been reported to be a putative cyanide hydratase based on sequence similarity (Reed et al., 2003) and to be a toxin protein of a toxin/antitoxin operon, which is the most induced gene in E. coli persister cells (Shah et al., 2006). Differential expression analysis by DNA microarrays indicated that MqsR is a global regulator of E. coli gene expression (González Barrios et al., 2006); for example, MqsR induces flhDC and lsrK and represses glpDFK (González Barrios et al., 2006), which further supports the model of quorum-sensing regulation of biofilm through MqsR. In addition, the recognition that MqsR induces ymgB (González Barrios et al., 2006) is consistent with the recent finding that this gene is critical for E. coli biofilm formation and acid resistance influenced by indole (Lee et al., 2007c). Two uncharacterized putative transcription regulators, yncC (b1450) and yiaG (b3555), were also identified as induced by MqsR (González Barrios et al., 2006). Also, yncC was identified as induced by deleting the AI-2 regulator genes lsrR and lsrK (Li et al., 2007). Here, we show that YncC is important for E. coli biofilm formation, that YncC inhibits colanic acid overproduction and thereby inhibits mucoidy by repressing expression of periplasmic protein YbiM, and that YncC plays a key role in the regulation of biofilm formation by quorum-sensing regulator MqsR.
Materials and methods
Bacterial strains, plasmids, media and growth conditions
The strains and plasmids used in this study are listed in Table 1. E. coli K12 MG1655 was obtained from the E. coli Genome Project of the University of Wisconsin-Madison (Blattner et al., 1997). E. coli K12 BW25113, its isogenic mutants and the pCA24N-based plasmids for expressing YncC, MqsR, B3023, LsrR, LsrK, LuxS and TqsA under the tight regulation via the lacIq repressor, were obtained from the Genome Analysis Project in Japan (Baba et al., 2006). Expression of the target proteins from these plasmids was induced by isopropyl-β-D-thiogalactopyranoside (IPTG) (Sigma, St Louis, MO, USA). The expression vector plasmid pBAD-Myc-His C (Invitrogen, Carlsbad, CA, USA) was used to construct pBAD-Myc-His C-b3023. Expression of C-terminal histidine-tagged B3023 protein under the arabinose-inducible araBAD promoter (PBAD) from this plasmid was induced by 0.5% L-arabinose (Acros Organics, Morris Plains, NJ, USA). The promoter-probe vector pPROBE-gfp[tagless] (Miller et al., 2000) was used to construct pPyncC-gfp, which carries the yncC promoter-gfp fusion.
Luria–Bertani medium (LB) was used to pre-culture all the E. coli cells (Sambrook et al., 1989) at 37 °C. LB medium was also used for bacterial growth rates, indole assays, green fluorescence protein (GFP) fluorescence intensity assays, the glass wool biofilm DNA microarrays and the nickel-enrichment DNA microarray experiments. To investigate the exopolysaccharide (EPS), bacterial strains were streaked on LB calcofluor plates (LB agar plates containing 100 μg ml−1 calcofluor (Sigma)) and also on LB Congo red plates (LB agar plate containing 20 μg ml−1 Congo red (Sigma) and 10 μg ml−1 Coomassie brilliant blue (Sigma)), incubated at 37 °C for 24–48 h and dye binding was evaluated by illuminating under UV light for calcofluor binding or by red color intensity for Congo red binding (Ferrieres et al., 2007).
Kanamycin (50 μg ml−1) was used for pre-culturing the E. coli BW25113 or MG1655 isogenic knockouts. Kanamycin (100 μg ml−1) was used for selecting plasmids pPROBE-gfp[tagless] and pPyncC-gfp. Chloramphenicol (30 μg ml−1) was used for selecting plasmid pCP20 and the plasmids derived from pCA24N. Both kanamycin (100 μg ml−1) and chloramphenicol (30 μg ml−1) were used to select for MG1655 hosting two plasmids, pPyncC-gfp and a pCA24N-based plasmids. Ampicillin (100 μg ml−1) was used for selecting plasmids pBAD-Myc-His C and pBAD-Myc-His C-b3023.
The specific growth rates of wild-type E. coli and the yncC mutant were determined by measuring the cell turbidity at 600 nm of two independent cultures of each strain as a function of time using values less than 0.7. Growth curves of the wild-type strain and the yncC mutant were obtained by measuring the cell turbidity at 600 nm of two independent cultures as a function of time until late stationary phase.
P1 transduction
Bacteriophage P1 transduction was used to construct mutants in MG1655 using BW25113-based mutants available from the KEIO collection (Baba et al., 2006). Briefly, to construct the MG1655 yncC ΩKmR mutant with a complete deletion of yncC (KmR: kanamycin-resistance marker gene), bacteriophage P1 was grown with BW25113 yncC ΩKmR, and the lysate was used to transduce MG1655. Kanamycin-resistance colonies were selected, and the correct yncC ΩKmR mutation in the MG1655 genome was confirmed by PCR using the forward locus-specific primer in the yncC upstream region 5′-GCAAAGTGGTGATCCGCG-3′ and the reverse kanamycin-specific primer K1, 5′-CAGTCATAGCCGAATAGCCT-3′. To construct the MG1655 yncC ybiM double mutation, the kanamycin-resistance gene was deleted from MG1655 yncC ΩKmR by introducing plasmid pCP20, which expresses the FLP recombinase to eliminate kanamycin-resistance selective marker (Cherepanov and Wackernagel, 1995), by curing the plasmid by 43 °C incubation and by selecting kanamycin-sensitive and chloramphenicol-sensitive colonies of MG1655 yncC KmR that lack pCP20 (Datsenko and Wanner, 2000). The ybiM ΩKmR mutation was then introduced into the MG1655 yncC KmR mutant from BW25113 ybiM of the KEIO collection (Baba et al., 2006) by bacteriophage P1 transduction, which led to the construction of the double mutant MG1655 yncC ybiM ΩKmR. The correct ybiM ΩKmR mutation in the MG1655 yncC ybiM ΩKmR genome was confirmed by PCR using the forward locus-specific primer in the ybiM upstream region, 5′-GCAAAGTGGTGATCCGCG-3′, and the reverse, kanamycin-specific primer K1. The double mutants MG1655 yncC wcaA ΩKmR, and yncC wcaD ΩKmR were constructed in the same way. The correct wcaA ΩKmR, and wcaD ΩKmR mutation in each double mutant was confirmed by PCR using a forward locus-specific primer, 5′-GGTCGTCATGTCGGAACCAC-3′ or 5′-CTCAATCAGATGGATGCGCT-3′, respectively, and the common reverse, kanamycin-specific primer K1. In a similar way, the MG1655 luxS KmR mutant was constructed by introducing the luxS ΩKmR mutation into the MG1655 wild-type strain from BW25113 luxS of the KEIO collection by bacteriophage P1 transduction to construct mutant MG1655 luxS ΩKmR, and the kanamycin-resistance gene was deleted from MG1655 luxS ΩKmR as described above.
Construction of plasmids
To construct a yncC promoter-gfp reporter fusion, a 323 bp DNA fragment containing the yncC promoter region (from 306 bp to 1 bp upstream of yncC start codon) was obtained by PCR using the BW25113 genome as the template and the pair of primers, 5′-CGCGGATCCGCGAAGAAATTACTGACGGT-3′ and 5′-GGGGAGCTCTTTAATTCCTTCGCGTGTTT-3′, which also inserted restriction sites, BamHI and SacI, upstream and downstream of the yncC promoter, respectively. After double digestion with restriction enzymes, BamHI and SacI, the DNA fragment was cloned into the promoter-probe vector plasmid pPROBE-gfp[tagless] (Miller et al., 2000), which led to the plasmid pPyncC-gfp.
To construct a plasmid expressing C-terminal histidine-tagged B3023 protein for the nickel-enrichment DNA microarrays, a 495 bp DNA fragment containing the b3023 gene was obtained by PCR using the BW25113 genome as a template and the pair of primers, 5′-AGCTCGAGAACAAACCTGACACTGGATGT-3′ and 5′-GTAAGCTTCCGCCAACGGCACATAAACAT-3′; this DNA fragment was cloned into expression vector pBAD-Myc-His C (Invitrogen), to form plasmid pBAD-Myc-His C-b3023, which expresses the C-terminal histidine-tagged B3023 under the L-arabinose-inducible PBAD promoter.
Crystal-violet biofilm assay
A static biofilm formation assay was performed in 96-well polystyrene plates as reported previously (Ren et al., 2005). Briefly, cells were inoculated with an initial turbidity at 600 nm of 0.05 in LB or LB glu at 37 °C or 30 °C for 24 h without shaking, then cell density (turbidity at 620 nm) and total biofilm (absorbance at 540 nm) were measured using crystal violet staining. Total biofilm (air/liquid and liquid/plastic interface) was normalized by bacterial growth for each strain. Each data point was averaged from at least 12 replicate wells (6 wells from each of the two independent cultures).
Indole assay
Extracellular, stationary-phase, planktonic indole concentrations of MG1655 and MG1655 yncC cultured in LB at 37 °C with shaking (250 r.p.m.) were measured spectrophotometrically as described previously (Domka et al., 2006) at 7, 15 and 24 h. Each experiment was performed with two independent cultures for each strain.
EPS and colanic acid assays
To quantify the total EPS, approximately 60 mg of mucoid or non-mucoid bacterial culture was collected from the surface of LB agar plates after a 12-h incubation at 37 °C, and then thoroughly resuspended in 1 ml sterile distilled water by vortex. The cell concentration was determined by cell turbidity at 600 nm. To inactivate EPS-degrading enzymes and to completely release EPS from the cell surface, the resuspended culture was boiled for 10 min. After cooling to room temperature, each sample was centrifuged at 16 000 g for 10 min, and the supernatant fraction was saved at −20 °C for quantification. The anthrone-H2SO4 assay was used to quantify total EPS as previously described based on quantification of glucose equivalents in the EPS (Mendrygal and González, 2000). Briefly, each 0.4 ml suitably-diluted sample was mixed with 0.1 ml of fresh 2% anthrone (Acros Organics) in ethyl acetate (Acros Organics) in a 10 ml glass test tube. One milliliter of 95–97% H2SO4 (Acros Organics) was added into each sample slowly, and the reactions were cooled for 10 min to room temperature, then the value of the absorbance at 620 nm of each reaction product was determined. Glucose equivalents in the EPS samples were quantified using a calibration curve with glucose (Sigma) from 10 to 80 μg ml−1, and the values were normalized by cell turbidity at 600 nm. All experiments were performed with two independent cultures.
Colanic acid from bacterial colonies was assayed by measuring fucose, the specific sugar component of this EPS (Obadia et al., 2007), according to the method of Obadia et al., (2007), which is based on the specific difference in absorbance at 396 and 427 nm after reacting fucose with sulfuric acid and cysteine hydrochloride. A L-fucose (Acros Organics) calibration curve was used (10–60 μg ml−1) to determine the fucose concentration, and these values were normalized by cell turbidity at 600 nm. All experiments were performed with two independent cultures. As a negative control, glucose was assayed and found to not produce a significant signal (as low as that using distilled water that lacked fucose); hence, the assay is specific to colanic acid.
P1 phage infection sensitivity assay
The bacteriophage infection sensitivity assay was performed as described before (Ho and Waldor, 2007) with the following modification. An overnight culture (12 h) was centrifuged (1500 g) for 5 min, and resuspended in LB to a turbidity of 3 at 600 nm. A serial of dilution of the P1 phage was prepared (100, 10−1, 10−2, 10−3, 10−4 and 10−5). A 0.01 ml volume of each serial dilution of the P1 phage was mixed with 0.1 ml of cell culture and incubated for 10 min at 37 °C. The cells were pelleted by centrifugation (1500 g) for 5 min to remove unattached phage, resuspended in 0.1 ml LB, mixed with 3 ml of the top agar (LB with 5 mM CaCl2, and 0.7% agar, kept at 50 °C) and then poured onto a standard LB plate. After solidification, the plates were incubated at 37 °C overnight, and then plaques were counted based on suitable phage dilution. Two independent cultures were used for all the experiments.
GFP fluorescence intensity assay
The specific GFP fluorescence intensity of cell cultures were found by diluting each culture quickly with phosphate-buffered saline (PBS, NaCl 8 g l−1, KCl 0.2 g l−1, Na2HPO4 1.44 g l−1 and KH2PO4 0.24 g l−1, pH 7.4) (Sambrook et al., 1989) in 96-well plates to a turbidity of 0.1–0.2 at 600 nm. The intensity of GFP fluorescence was quantified with a Spectra Max Germini EM fluorescence microplate reader (Molecular Devices, Sunnyvale, CA, USA) using an excitation wavelength of 485 nm, an emission wavelength of 528 nm and a cutoff wavelength of 515 nm. The cell turbidity was quantified at 600 nm with a Sunrise microplate reader (Tecan, Austria Gesellschaft, Salzburg, Austria), and the GFP fluorescence intensity of each of the cell suspensions was normalized by its cell density. The promoter-less GFP strain MG1655/pPROBE-gfp[tagless] was used as the negative control. Two independent cultures were used for the experiments.
Total RNA isolation and DNA microarrays
DNA microarray experiments were performed to examine differential gene expression in biofilm cells upon deleting yncC in two independent ways as follows: (i) E. coli K-12 MG1655 yncC colony cells were grown on LB agar plates at 37 °C for 15 h as there was a clear phenotype of mucoidy vs the wild-type strain, and (ii) E. coli BW25113 yncC biofilm cells were developed on glass wool at 37 °C for 15 h. For the agar biofilm, fresh single colonies of wild-type E. coli MG1655 and MG1655 yncC were re-streaked on LB agar plates, incubated and about 0.05 g of the colony cultures on an LB plate were quickly transferred to 2-ml collection tubes. The total RNA was isolated from these colony cells as described previously (Ren et al., 2004a). The E. coli GeneChip Genome 2.0 Array (Affymetrix, P/N 900551, Santa Clara, CA, USA) containing 10 208 probe sets for open reading frames, rRNA, tRNA and intergenic (IG) regions for four E. coli strains: MG1655, CFT073, O157:H7-Sakai and O157:H7-EDL933, was used to analyze the complete E. coli transcriptome.
For the glass wool biofilm, overnight cultures (16 h, 2.5 ml) of wild-type E. coli BW25113 and BW25113 yncC in LB and LB with kanamycin (50 μg ml−1), respectively, were used to inoculate 250 ml LB with 10 g of glass wool (Corning Glass Works, Corning, NY, USA) for forming biofilm (Ren et al., 2004a). After incubating at 37 °C for 15 h with shaking (250 r.p.m.), biofilm cells were prepared by rinsing and sonicating the glass wool in sterile 0.85% NaCl solution at 0 °C as described before (Zhang et al., 2007). The total RNA was isolated from biofilm cells as described previously (Ren et al., 2004a). The E. coli Genechip antisense genome array (P/N 900381, Affymetrix) containing probes for more than 4200 open reading frames was used to analyze the complete E. coli transcriptome as described previously (Zhang et al., 2007).
For both sets of microarrays, hybridization was performed for 16 h, and the total cell intensity was scaled to an average value of 500. The probe array images were inspected for any image artifact. Background values, noise values and scaling factors of both arrays were examined and were comparable. The intensities of polyadenosine RNA control were used to monitor the labeling process. As expected, signals of the deleted genes, araA and rhaA, were low for both BW25113 and BW25113 yncC, whereas the signals of yncC were low for the yncC mutants for both BW25113 and MG1655. For both sets of binary microarray comparisons to determine differential genes expression, if the gene with the larger transcription rate did not have a consistent transcription rate based on the 11–15 probe pairs (P-value less than 0.05), these genes were discarded. A gene was considered differentially expressed when the P-value and the corrected P-value based on the False Discovery Rate Method (Benjamini and Hochberg, 1995) for comparing two chips were lower than 0.05 (to assure that the change in gene expression was statistically significant and that false positives arise less than 5%) and when the expression ratio was higher than the s.d. for all of the E. coli K-12 genes (1.6-fold for both) (Ren et al., 2004b). The gene functions were obtained from the National Center for Biotechnology Information database (Wheeler et al., 2006), and the EcoCyc database (Keseler et al., 2005).
Nickel-enrichment DNA microarrays
Overnight cultures of E. coli K-12 BW25113 carrying pBAD-Myc-His C or pBAD-Myc-His C-b3023 cells were diluted (1:100) into 250 ml LB with ampicillin (100 μg ml−1). The cultures were grown to a turbidity of 0.8 at 600 nm at 37 °C with shaking (250 r.p.m.), and then were kept at 4 °C for 45 min to stop growth. L-arabinose (final concentration of 0.5%) was added to induce the synthesis of C-terminal histidine-tagged B3023 protein from pBAD-Myc-His C-b3023. The cultures were incubated overnight at room temperature with shaking (160 r.p.m.). Formaldehyde (1%, Fisher, Fair Lawn, NJ, USA) was applied to promote crosslinking between the C-terminal histidine-tagged B3023 protein and the DNA to which it was bound (Tamimi et al., 2004), and the cultures were incubated for another 20 min with shaking (100 r.p.m.) at room temperature. Glycine (0.125 M, Sigma) was applied to stop the crosslinking for 5 min with shaking (100 r.p.m.). The cells were collected, washed with 200 ml of cold sodium chloride-phosphate (0.5 M NaCl, 50 mM NaH2PO4, pH 8) buffer and resuspended in 25 ml of the same buffer with 1 mM phenylmethylsulfonyl fluoride (Pierce Biotechnology, Rockford, IL, USA). The cells were lysed by a French press (Thermo Electron Corp., San Jose, CA, USA), and the lysate was centrifuged at 12 000 g force at 4 °C for 20 min to remove the cellular debris. Ni-NTA agarose gel resin was used to bind the C-terminal His6-tagged B3023-DNA complexes. The eluate with B3023-DNA complexes was treated with 75 μg ml−1 RNase A and 100 μg ml−1 proteinase K. The RNA and protein-free DNA fragments were then purified, and the presence of DNA fragments of 100–1000 bp was confirmed by gel electrophoresis. The DNA fragments were labeled with biotin at 3′-end, and the protein/RNA-free DNA samples were prepared for microarray analysis as described previously (Lee et al., 2007c). The DNA fragments were labeled and DNA microarrays were performed using E. coli Genechip antisense genome array (P/N 900381, Affymetrix) (Ren et al., 2004a). For analysis of the data, we chose as positive candidate IG regions with a signal greater than 1000 and at least eightfold enriched compared to the non-B3023 control array (empty vector pBAD-Myc-His C) as the global average signal of all the genes and IG regions in the B3023 chip was 588 and the s.d. of the enrichment (B3023 chip signal compared to the empty vector signal) was 5.
Electrophoretic mobility shift assay
Electrophoretic mobility shift assays (EMSA) were performed as described previously (Zhang et al., 2007). To identify the promoter regions YncC binds, YncC was synthesized using the EasyXpress Linear Template Kit Plus (Qiagen, Valencia, CA, USA). Promoters lie at a variable distance upstream of their associated start codons, but in a well-defined window upstream of the gene transcription start sites (Gordon et al., 2006). In this work, we choose the region 200–300 bp upstream of the start codon of the associated genes as the promoter region, and five DNA fragments of these regions were amplified by PCR from genomic DNA of wild-type BW25113. The ybiM promoter region (262 bp, consisting of 233 bp of upstream and 29 bp downstream of the start codon of ybiM) was amplified by PCR with the primers 5′-CGACAGGGTAAATATCTG-3′ and 5′-GCAATCAGTAGTGTGAGG-3′. The ybeL promoter region (243 bp, consisting of 205 bp of upstream and 38 bp downstream of the start codon of ybeL) was amplified by PCR with primers 5′-CTACAAATGTAGCGTTGA-3′ and 5′-GCAACCAGTTCACGGTAA-3′. The yebT promoter region (262 bp, consisting of 225 bp of upstream and 37 bp downstream of the start codon of yebT) was amplified by PCR with primers 5′-GCCAGCAAGGTTTACGCA-3′ and 5′-GCGCTTCAGTCGTCGAAG-3′. The sfsA promoter region (301 bp, consisting of 253 bp of upstream and 48 bp downstream of the start codon of sfsA) was amplified by PCR with primers 5′-GCTTAATCCAGCTGGCGA-3′ and 5′-GTAACGCTGAATTAGCGT-3′. The uspF promoter region (272 bp, consisting of 231 bp of upstream and 41 bp downstream of the start codon of uspF) was amplified by PCR with primers 5′-CAACCTGTTGGATGAAGA-3′ and 5′-GAATCGGAAATATCGATA-3′. The PCR products were gel purified with a QIAquick Gel Extraction Kit (Qiagen) and then labeled with biotin using the Biotin 3′-end DNA Labeling Kit (Pierce Biotechnology). For the binding reaction, in vitro-synthesized YncC (0.5 μl out of total 25 μl product in the in vitro-translation system) was incubated with biotin-labeled target promoter (8 ng) and the nonspecific competitor DNA (poly dI·dC, 1 μg) in a 20 μl binding reaction system supplied by the EMSA kit at room temperature for 15 min. For the competition assay, unlabeled promoter regions from 200 ng to 1 200 ng were used to confirm the specificity of protein-DNA binding. After the binding reaction, electrophoresis was conducted at 100 V for 2.5 h at 4 °C using a 6% DNA retardation gel (Invitrogen), then the samples were transferred to a nylon membrane (Roche Diagnostics GmbH, Mannheim, Germany) using a Mini Trans-Blot Electrophoretic Transfer Cell (Bio-Rad Laboratories, Hercules, CA, USA), and 3′-biotin-labeled DNA was detected with the LightShift Chemiluminescent EMSA kit (Pierce Biotechnology). Each experiment was performed twice.
Similarly, to identify the promoter regions B3023 protein binds, B3023 was synthesized. Four DNA fragments of promoter regions, the fragment with the yncC promoter (IG953), the fragment with the yiaT and yiaU promoters (IG2314), the fragment with the ssrS promoter (IG1895) and the fragment with the rybA and mntR promoters (IG543), were amplified by PCR from genomic DNA of the wild-type strain BW25113. The yncC promoter region (278 bp, consisting of 232 bp of upstream and 46 bp downstream of the start codon of yncC) was amplified by PCR with the primers 5′-GCAAAGTGGTGATCCGCG-3′ and 5′-CCTGCAGAGTCAAACTGA-3′. The yiaT and yiaU promoter regions (242 bp, consisting of 180 bp of upstream and 62 bp downstream of the start codon of yiaT) was amplified by PCR with primers 5′-GCAGTTGCGCTTGCCATA-3′ and 5′-GGCAGCGATTACCGAGAT-3′. The small RNA ssrS promoter region (279 bp, 3, 053, 876–3, 054, 154 on the E. coli K-12 chromosome) (Keseler et al., 2005) was amplified by PCR with primers 5′-GCGTATTCGGATGCTGCA-3′ and 5′-CCTTGAACCCTTGGTTCA-3′. The small RNA rybA and mntR promoter region (263 bp, 852, 154–852, 416) was amplified by PCR with primers 5′-CAGCATTAACCGTACTTT-3′ and 5′-GCGACGACTCATTGTGCT-3′.
Microarray data accession number
The expression data have been deposited in the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) and are accessible through GEO series accession number GSE8706 (Edgar et al., 2002).
Results
Deletion of yncC decreases biofilm formation
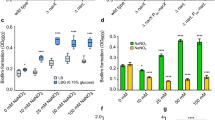
To investigate whether YncC controls biofilm formation, biofilm formation of the yncC deletion mutant was measured using the 96-well crystal-violet assay in LB at 37 °C. Growth-normalized biofilm formation of MG1655 yncC decreased 2.2±0.5-fold in LB medium at 37 °C after 24 h compared to that of wild-type MG1655 (Figure 1). This biofilm defect was complemented by expressing yncC from plasmid pCA24N-yncC in MG1655 yncC using 0.1 mM IPTG since biofilm formation increased significantly (Figure 1). In addition, on deleting yncC, biofilm formation was reduced 1.9±0.4-fold in LB glu medium (LB medium supplemented with 0.2 % (m/v) glucose) at 37 °C, was reduced 2.3±0.5-fold in LB glu at 30 °C and was reduced 1.7±0.3-fold in LB at 30 °C (Figure 1). As biofilm formation is dynamic, we examined its formation as a function of time on deleting yncC, and found biofilm decreased 2.5±0.3-, 3.0±0.4-, 2.2±0.3- and 1.8±0.3-fold in LB medium at 37 °C after 7, 15, 24 and 48 h of incubation, respectively.

Total biofilm (air/liquid and liquid/plastic interface) normalized by bacterial growth (turbidity at 620 nm) after 24 h of incubation in LB or LB glu at 37 or 30 °C for the wild-type MG1655 and the mutant MG1655 yncC strains. Each experiment was performed in duplicate, and one s.d. is shown. LB, Luria–Bertani medium.
Deletion of yncC increases extracellular indole
On deleting yncC in MG1655, the extracellular concentration of the biofilm signal indole increased 1.7±0.2-, 2.1±0.1- and 1.5±0.2-fold after 7, 15 and 24 h in LB medium at 37 °C, respectively. Since indole acts as an extracellular signal that represses biofilm formation in E. coli K-12 (Domka et al., 2006; Lee et al., 2007b), the increase of extracellular indole concentration by deleting yncC is consistent with the observation that the yncC mutation decreased biofilm formation in LB medium.
Deletion of yncC elicits mucoidy
The MG1655 yncC culture became noticeably mucoid compared to the wild-type strain on LB agar (Figure 2a). As excessive production of bacterial EPS is a common cause of mucoidy in E. coli and other Gram-negative bacteria (Zinkewich-Peotti and Fraser, 1988; Qiu et al., 2007), we collected EPS from mucoid MG1655 yncC culture and evaluated bacterial EPS production using an anthrone-H2SO4 assay. The normalized EPS from the mucoid MG1655 yncC culture was 10±4-fold higher than that from the non-mucoid wild-type MG1655 culture. Therefore, yncC mutation leads to EPS overproduction and elicits mucoidy.

Escherichia coli MG1655 mutant yncC ΩKmR and MG1655 yncC ΔKmR cultures exhibit mucoidy on Luria–Bertani (LB) agar plates after 16 h of incubation at 37 °C, whereas the wild-type MG1655 and the double mutant MG1655 yncC ybiM ΩKmR cultures lack mucoidy (a). E. coli MG1655 yncC ΔKmR cultures exhibit mucoidy on LB agar plates after 16 h incubation at 37 °C, whereas wild-type MG1655 and the double mutants MG1655 yncC wcaA ΩKmR and MG1655 yncC wcaD ΩKmR cultures lack mucoidy (b).
We also noticed that bacteriophage P1-mediated transduction using the strain MG1655 carrying the yncC mutation had a dramatically lower efficiency compared to using wild-type MG1655; deleting yncC from MG1655 increased bacteriophage P1 resistance 52±16-fold compared to the wild-type MG1655. This result confirms that YncC represses EPS production as EPS overproduction increases bacteriophage P1 resistance by masking phage receptors on the bacterial surface thereby blocking phage infection (Petty et al., 2007).
Mucoidy is from overproduction of colanic acid
As colanic acid is an EPS that is commonly found in E. coli (Stevenson et al., 1996), and as we have found its synthesis is controlled in a temporal manner in biofilms (Domka et al., 2007) and is related to AI-2 signaling (Herzberg et al., 2006), we investigated colanic acid production of MG1655 yncC mutant and the wild-type MG1655 strain by measuring fucose, which is a specific component of this EPS (Obadia et al., 2007). Under the same conditions, the normalized colanic acid from the mucoid MG1655 yncC colonies was 326±173-fold higher than that from the non-mucoid MG1655 colonies. Therefore, colanic acid is the overproduced EPS caused by the yncC mutation, and YncC inhibits E. coli colanic acid overproduction and thereby represses mucoidy.
To confirm these results, we deleted the colanic acid generation genes wcaA and wcaD along with yncC from MG1655 and investigated mucoidy in for the double mutants wcaA yncC and wcaD yncC. The deletion of either colanic acid gene dramatically repressed the MG1655 yncC mucoidy phenotype (Figure 2b); hence, YncC inhibits colanic acid overproduction.
To see if other types of EPS are overproduced by deleting yncC, EPS-calcofluor and EPS-Congo red binding tests were performed. Calcofluor as a fluorochrome binds to (1 → 3)-β- and (1 → 4)-β-D-glucopyranosides such as those found in cellulose, chitin and succinoglycans (Da Re and Ghigo, 2006). Congo red, along with binding curli, also binds (1 → 4)-α-D-glucopyranosides and basic or neutral EPS (Ferrieres et al., 2007). However, both MG1655 yncC and wild-type MG1655 failed to bind calcofluor, which is consistent with previous reports for some other E. coli K12 MG1655 strains (Da Re and Ghigo, 2006), and both MG1655 yncC and wild-type MG1655 had the same Congo red-staining levels. These observations suggest that only colanic acid was overproduced by deleting yncC, whereas other EPS, such as cellulose, chitin, succinoglycans, Congo red-binding basic EPS or neutral EPS, were not overproduced by deleting yncC.
Genes regulated by YncC in biofilms
Whole-transcriptome analysis indicated that deletion of yncC induced 30 genes (Table 2) and repressed 10 genes more than fourfold in glass wool biofilm cells (Table 3). Among the most induced genes, ptsH (4.0-fold) encodes a heat-stable histidyl phosphorylatable protein (Postma et al., 1993) that is induced by BssR/BssS in biofilms (Domka et al., 2006). Also induced was cpxP (4.6-fold) that encodes a periplasmic adaptor protein involved in resistance to extracytoplasmic stress (Danese and Silhavy, 1998), which was induced in E. coli TG1 and TG biofilms (Beloin et al., 2004) and is involved in adhesion of E. coli (Otto and Silhavy, 2002). sixA was induced 4.0-fold and encodes a signal inhibitory factor-X (Ogino et al., 1998) that was induced in E. coli TG1 and TG biofilms (Beloin et al., 2004). fldA was induced 5.3-fold and encodes flavodoxin 1 (Puan et al., 2005) that was induced in the human urine biofilms of the asymptomatic E. coli bacteriuria strains (Hancock and Klemm, 2007).
Several poorly characterized genes were also induced on deletion of yncC including ynhG (4.6-fold), which was repressed in E. coli MG1655 biofilms (Schembri et al., 2003), ybiM (6.5-fold), which was induced by AI-2 in E. coli W3110 (DeLisa et al., 2001b), yqjC (4.6-fold), which was repressed by AI-2 in E. coli W3110 (DeLisa et al., 2001b) and yahO (5.7-fold), which was induced by MqsR expression in E. coli MG1655 (González Barrios et al., 2006).
Among the most repressed genes on deletion of yncC in biofilm cells (Table 3), yceK was repressed 4.0-fold and encodes a lipoprotein that is repressed in E. coli JM109 biofilms (Ren et al., 2004a). Also repressed was motB (4.6-fold) that encodes a flagellar motor rotation protein whose mutation causes severe defects in biofilm formation (Pratt and Kolter, 1998). yebT was repressed 5.6-fold and encodes a putative outer membrane protein that was induced previously by LuxS (Wang et al., 2005). Other highly-repressed genes include hyfD (4.3-fold), which encodes a hydrogenase membrane subunit (Andrews et al., 1997), frlA (3.0-fold), which encodes a fructoselysine transporter (Wiame et al., 2002) and yieL (7.0-fold), which encodes a putative xylanase; all three loci are repressed in LB glu biofilms by the AI-2 exporter TqsA (Herzberg et al., 2006). Finding differential expression of these biofilm-related genes on deletion of yncC supports that YncC plays an important role in the regulation of E. coli biofilm formation.
Seven of the most-induced genes on deleting yncC in biofilm cells were also induced on deleting yncC in colony cells from LB agar plates based on an independent set of whole-transcriptome analysis (Table 2). The seven genes are as follows: (i) sfsA, encoding a predicted maltose metabolism transcriptional regulator, (ii) yfiA, encoding the stationary-phase translation inhibitor and ribosome stability factor, (iii) rpsV, encoding the 30S ribosomal subunit protein, (iv) dppA, encoding the dipeptide transport protein and (v, vi, vii) three genes with unknown function, ybiM, ydiZ and yjdN. An insertion sequence element-interrupted Rac prophage lom homolog gene, lomR_1, was the only repressed gene identified by both sets of microarrays (Table 3). As none of the previously-characterized colanic acid generation genes were induced on deleting yncC in either set of DNA microarrays, YncC may regulate colanic acid production not at the transcription level but instead through an unknown indirect pathway.
YncC represses ybiM
The YncC protein sequence is conserved in E. coli strains, since BLAST analysis indicated that the 221 amino acid (aa) E. coli K-12 YncC shares 220 identical aa with the E. coli O157:H7 YncC (also 221 aa). Structure prediction analysis of YncC using ExPASy server (Gasteiger et al., 2003) suggested that YncC is a putative DNA-binding transcription regulator with a helix-turn-helix DNA-binding domain (aa 37–56). We suspected YncC directly binds to the promoter regions of some of the genes identified by the DNA microarrays and functions as an inducer or repressor to direct expression of these genes. Therefore, YncC binding of the promoter regions of the two most-induced genes in both glass wool and agar biofilm cells (ybiM and sfsA, Table 2), the two most-induced genes in glass wool biofilm cells (ybeL and uspF, Table 2), as well as with the one of the most repressed genes in glass wool biofilm cells (yebT, Table 3) was investigated using gel shift experiments. We were especially interested in ybiM and yebT since not only were they differentially expressed on yncC mutation but also both of them are regulated by AI-2 (DeLisa et al., 2001b; Wang et al., 2005). It was found that YncC binds to the promoter region of the ybiM gene (Figure 3a), but YncC did not bind under these conditions to the promoter regions of ybeL, sfsA, uspF and yebT (Figure 3b and data not shown). As DNA microarrays show that ybiM expression is induced 6.5-fold on deleting yncC (Table 2), this indicates that the putative transcription regulator YncC binds as a repressor of ybiM, which encodes a putative periplasmic protein with no known function.

Electrophoretic mobility shift assay (EMSA) to test the binding of YncC to the ybiM promoter (a) and to the ybeL promoter (b), and binding of B3023 to the ybeL promoter (c) and to the IG1895 region (d). Lane 1: labeled promoter, lane 2: protein and labeled promoter, lane 3: protein, labeled promoter, and unlabeled promoter and lane 4: protein, labeled promoter, and increased unlabeled promoter. The triangle indicates that increasing amounts of unlabeled promoter were used as a specific competitor of the DNA–protein interaction. Each experiment was performed twice.
YncC regulates biofilm formation through ybiM
The poorly characterized YbiM belongs to the YhcN family of periplasmic proteins that includes BhsA; we found BhsA influences biofilm formation through hydrophobicity and stress response (Zhang et al., 2007). In addition, earlier microarray analyses indicated that ybiM was induced 4.7-fold by the quorum-sensing signal AI-2 in E. coli K12 W3110 (DeLisa et al., 2001b).
To investigate whether YncC regulates biofilm formation through ybiM, we constructed a MG1655 yncC ybiM double mutant. Biofilm formation of the MG1655 yncC ybiM double mutant in LB medium at 37 °C increased 2.3±0.7-fold compared to the MG1655 yncC mutant. This indicates that the ybiM mutation represses the reduction seen in biofilm formation on deletion of yncC, which suggests that YncC influences biofilm formation most likely by regulating ybiM expression. In addition, the MG1655 yncC ybiM double mutant demonstrates the non-mucoid appearance of wild-type MG1655 (Figure 2a), which indicates again that the ybiM mutation represses phenotype seen with the yncC mutation. Consistently, colanic acid production of the MG1655 yncC ybiM double mutant was as low as the wild-type strain under the same conditions; that is, colanic acid production of the MG1655 yncC ybiM double mutant was decreased 173±65-fold compared to the MG1655 yncC mutant. Corroborating the reduction in colanic acid, sensitivity to bacteriophage P1 infection for the MG1655 yncC ybiM double mutant increased 62±27-fold compared to the MG1655 yncC mutant; hence, the double mutant was as sensitive to P1 as the wild-type strain. All these results indicate that YncC functions as the ybiM repressor and elicits its effect on mucoidy and biofilm formation by regulating ybiM.
yncC expression is influenced by MqsR, LsrR, LsrK, TqsA and YncC
We also investigated the possibility that AI-2 regulates yncC expression as previous microarray analysis indicated that the E. coli quorum-sensing regulator MqsR induces yncC expression (González Barrios et al., 2006). The effect of MqsR on yncC expression was quantified with a yncC promoter-gfp fusion (pPyncC-gfp) while overexpressing MqsR from pCA24N-mqsR. The specific GFP fluorescence intensity of MG1655/pPyncC-gfp/pCA24N-mqsR with 0.2 mM IPTG to induce mqsR increased 1.4±0.2-fold compared to that without IPTG induction and increased 1.7±0.2-fold with 0.5 mM IPTG induction (Figure 4); hence, MqsR induces yncC promoter activity, which is consistent with our previous microarray analysis that showed yncC expression is repressed on deleting mqsR (González Barrios et al., 2006).

The GFP fluorescence intensity normalized with cell turbidity at 600 nm of MG1655/pPyncC-gfp overexpressing MqsR, B3023, TqsA, LsrR, LsrK or LuxS with IPTG induction from plasmid pCA24N-mqsR, pCA24N-b3023, pCA24N-tqsA, pCA24N-lsrR, pCA24N-lsrK or pCA24N-luxS, respectively. The experiments were repeated at least two times (one representative data set shown), and one s.d. is shown. GFP, green fluorescent protein; IPTG, isopropyl-β-D-thiogalactopyranoside.
In a similar manner, we evaluated the effect of several other AI-2-related proteins on yncC expression, including LuxS (the AI-2 synthesis protein), LsrR (the repressor of the AI-2 uptake transporter complex), LsrK (cytoplasmic kinase, which phosphorylates AI-2 into an activated molecule inside E. coli cells) and TqsA (exporter of AI-2) (Figure 4). Adding 0.5 mM IPTG increased the specific GFP fluorescence intensity of MG1655/pPyncC-gfp/pCA24N-lsrR 2.7±0.7-fold, increased the value of MG1655/pPyncC-gfp/pCA24N-lsrK 2.3±0.5-fold, increased the value of MG1655/pPyncC-gfp/pCA24N-tqsA 1.5±0.4-fold and decreased the value of MG1655/pPyncC-gfp/pCA24N-luxS 2.5±0.6-fold (Figure 4). As a control, the presence of IPTG did not affect the value of the specific GFP fluorescence intensity of MG1655/pPyncC-gfp/pCA24N (empty vector control) (Figure 4). As additional negative controls, under the same test conditions, adding 0.5 mM IPTG to induce protein CRP or Crl did not significantly affect the value of the specific GFP fluorescence of the strain MG1655/pPyncC-gfp/pCA24N-crp or MG1655/pPyncC-gfp/pCA24N-crl (data not shown). Like YncC, both CRP and Crl are transcription regulatory proteins. These results indicate that LsrR, LsrK and TqsA induce yncC expression, whereas LuxS represses yncC expression.
The yncC promoter activities of wild-type E. coli MG1655 and the E. coli MG1655 yncC KmR mutant were also evaluated by measuring the specific GFP fluorescence intensities of MG1655/pPyncC-gfp and MG1655 yncC KmR/pPyncC-gfp. Under the same conditions, the specific GFP fluorescence intensity of MG1655 yncC KmR/pPyncC-gfp was 2.0±0.3-fold higher than that of MG1655/pPyncC-gfp, which indicates that YncC represses its own promoter activity.
Putative transcription regulator B3023 enhances MqsR toxicity and represses yncC
The quorum-sensing protein MqsR is a global regulator in E. coli (González Barrios et al., 2006) and also elicits a toxic effect on E. coli growth (Shah et al., 2006). We suspected that the upstream gene of mqsR (b3022), b3023, a putative transcription regulator, may be involved in regulation with MqsR so we examined the effect of deleting b3023 on the toxicity of MqsR by overexpressing MqsR via IPTG induction from plasmid pCA24N-mqsR in both E. coli MG1655 b3023 and wild-type MG1655. Without overexpressing MqsR, MG1655 b3023/pCA24N (empty vector) and MG1655/pCA24N exhibit similar growth (Figure 5); however, overexpressing MqsR with 2 mM IPTG decreases the growth of wild-type MG1655/pCA24N-mqsR by 9.3±1.0-fold after 5 h (Figure 5). This confirms that MqsR has a toxic effect on MG1655. When overexpressing MqsR with 2 mM IPTG under the same conditions, the growth of MG1655 b3023/pCA24N-mqsR was only decreased by 2.2±0.2-fold after 5 h, so the decrease was significantly reduced compared to MG1655/pCA24N-mqsR with 2 mM IPTG (Figure 5). This indicates that the b3023 mutation reduces the toxic effect of MqsR, which also supports our hypothesis that the putative transcription regulator B3023 may be involved in the MqsR (B3022) regulation pathway. This was further supported by the finding that deleting b3023 in E. coli K-12 BW25113 genetic background also similarly represses the toxic effect of MqsR (data not shown).

Impact of the Δb3023 mutation on the bacterial growth of MG1655 while overexpressing MqsR. MqsR was expressed from plasmid pCA24N-mqsR with 2 mM IPTG induction. Each experiment was performed in duplicate, and one s.d. is shown. IPTG, isopropyl-β-D-thiogalactopyranoside.
To determine what promoters B3023 binds in vivo, we used nickel-enrichment DNA microarrays and identified 16 IG regions that the putative transcription regulator B3023 may bind (Table 4). These results indicate that B3023 binds IG953 that includes the yncC gene upstream promoter region (enriched 15-fold); therefore, B3023 appears to regulate yncC expression. Interestingly, the 16 IG regions also include three regions covering small non-coding RNA (sRNA) and their upstream promoter regions: IG1895 covering sRNA ssrS (enriched 16-fold), which represses expression from sigma70-dependent promoters in the stationary phase (Wassarman and Storz, 2000), IG543 covering sRNA rybA (enriched 9-fold) and IG2089 covering sRNA ryhA (enriched 9-fold); rybA and ryhA, like other sRNA, may play important roles as regulators of translation and message stability (Gottesman, 2004).
We next tried to confirm the in vitro binding of B3023 with four IG regions by gel shift experiments including IG953 (including the yncC promoter), IG2314 (including the yiaT and yiaU promoters that were enriched 20-fold) and two sRNA IG regions (IG543 including sRNA rybA, and IG1895 including sRNA ssrS). Gel shift experiments clearly indicated that B3023 specifically binds to the yncC promoter under our test conditions (Figure 3c), but does not bind to the DNA fragments of the other three IG regions, IG2314, IG1895 and IG543, under the same conditions (Figure 3d and data not shown). This indicates that yncC expression is under the control of putative transcription regulator B3023, as predicted by the nickel-enrichment DNA microarrays.
To determine if the binding of B3023 regulates yncC expression as a repressor or inducer, we quantified E. coli MG1655 yncC promoter activity with specific GFP fluorescence intensity with a yncC promoter-gfp fusion via plasmid pPyncC-gfp while overexpressing B3023 via plasmid pCA24N-b3023 with IPTG induction. The specific GFP fluorescence intensity of MG1655/pPyncC-gfp/pCA24N-b3023 cells with 0.5 mM IPTG to induce B3023 decreased 1.5±0.3-fold compared to that without IPTG induction and decreased 2.5±0.7-fold with 2 mM IPTG induction (Figure 4). Hence, B3023 represses yncC.
Discussion
Previous research showed the cross-species bacterial communication quorum-sensing signal AI-2 stimulates E. coli biofilm by stimulating motility genes, and this effect is mediated by MqsR, which then regulates QseBC (González Barrios et al., 2006). In this study, by investigating the biofilm changes caused by mutating the transcription regulator YncC, by investigating the genes regulated by YncC via DNA microarrays, by investigating which promoters are directly bound by YncC using gel shift experiments and by investigating which proteins affect yncC expression, we demonstrate that MqsR-regulated YncC influences biofilm formation and mucoidy by repressing expression of the periplasmic protein YbiM. We also demonstrate B3023 regulates yncC as a repressor.
YbiM belongs to the YhcN family, which has nine paralogous, low-molecular-weight proteins: BhsA (Zhang et al., 2007), YahO, YbiJ, YbiM, YdgH, YhcN, YjfN, YjfO and YjfY in E. coli (Rudd et al., 1998). Sharing a predicted signal peptide in the N-terminus and a common motif in the C-terminus with unknown function (TAXIYK, where A and Y are conserved for all nine proteins and X represents any aa), the YhcN family of proteins were proposed to interact functionally and/or structurally with each other, to have evolved from a common ancestor, and to play roles in self-identification or colony organization by cell-cell contact or intercellular signaling (Rudd et al., 1998). We demonstrated that one of the YhcN family proteins, BhsA (YcfR), influences biofilm formation through hydrophobicity and stress response (Zhang et al., 2007). Here, we demonstrate that another YhcN family member, YbiM, is regulated by YncC, and that YbiM affects both biofilm formation and mucoidy. Apart from BhsA and YbiM, the functions of the other members of this family remain unknown, so whether they are related to biofilm formation deserves to be investigated as a means to discover the role of this protein family in biofilm development.
E. coli produces different types of EPS including serotype-specific lipopolysaccharide O-antigen, capsular K-antigen, colanic acid, (1 → 4)-β-glucan cellulose and (1 → 6)-β-N-acetylglucosamine polymer, which play important roles in the interaction between the bacterium and its environment (Ferrieres et al., 2007). The precise role of the EPS in bacteria is dependent on the natural environment of the microorganism (Kumar et al., 2007). In E. coli, overexpressing the genes involved in colanic acid synthesis, such as djlA and rcsF, induces colanic acid overproduction and elicits mucoidy (Genevaux et al., 2001; Potrykus and Wegrzyn, 2004). Here, we demonstrate that the yncC mutation leads to overproduction of colanic acid and so elicits mucoidy, and at the same time represses biofilm formation. Colanic acid is critical for the formation of the complex three-dimensional structure and depth of E. coli biofilms, while is not required for surface attachment (Danese et al., 2000). In contrast, recent research indicates that overproduced E. coli colanic acid blocks the time-dependent adhesion of bacteria to both hydrophilic and hydrophobic surfaces (Hanna et al., 2003). So, most likely the yncC mutation represses E. coli biofilm formation at the early stage by abnormally overproducing colanic acid, which blocks bacterial attachment. Consistent with our results, the quorum-sensing-impaired mutant of the phytopathogenic bacterium Pantoea stewartii subsp. stewartii elicits hypermucoidy and EPS-overproduction with the result that it attaches poorly to surfaces and forms poor biofilms (Koutsoudis et al., 2006).
As EPS can mask bacteriophage receptors on bacterial surfaces to block phage infection (Forde and Fitzgerald, 2003), we suspected that the overproduction of colanic acid caused by the yncC mutation may also mask unknown signal receptors on the bacterial cell surface to block bacterial cell communication and then biofilm formation. Since deleting yncC did not cause differential expression of previously characterized colanic acid generation genes, yncC may regulate colanic acid production not at the transcription level but instead through an unknown indirect pathway that includes the periplasmic protein YbiM and/or other structural proteins on the bacterial cell surface.
E. coli MqsR is a small protein (98 aa), which has homologs in several other genera including Yersinia, Cupriavidus, Burkholderia, Xylella, Bordetella, Polaromonas, Pseudomonas, Nitrosospira and Ralstonia based on sequence similarity. However, MqsR has only been characterized in E. coli. In E. coli, MqsR is a global regulator of gene expression (González Barrios et al., 2006) and is a toxin protein (Shah et al., 2006; Figure 5), which has been suggested to selectively target DNA gyrase or topoisomerase (Shah et al., 2006). This raises the possibility that MqsR may elicit its global regulation effect on gene expression by affecting bacterial DNA tertiary structure. The genes regulated by MqsR include lsrK, encoding a cytoplasmic kinase that phosphorylates AI-2 into an activated molecule inside E. coli cells, and glpDFK, encoding a glycerol uptake and metabolism system that regulates AI-2 uptake and lsr operon transcription through cyclic AMP-catabolite activator protein-dependent regulation (Taga et al., 2003; Xavier and Bassler, 2005; González Barrios et al., 2006). This suggests that signal AI-2 not only stimulates biofilm formation through MqsR as indicated before (González Barrios et al., 2006), but also that AI-2 uptake and internalization is regulated by MqsR. Since no DNA-binding transcription regulator domain or other previous well characterized functional domain was predicted for MqsR, this suggests MqsR regulates the expression of genes, including these AI-2 related lsrK and glpDFK genes, through an unknown mechanism. The putative transcription regulator YncC we characterized here most likely is involved in this process.
We investigated the effect of the quorum-sensing signal AI-2 on yncC expression; however, under our test conditions, addition of exogenous AI-2 (Omm Scientific Inc., Dallas, TX, USA) did not significantly change yncC expression in MG1655 or in the MG1655 luxS KmR mutant. In contrast, overexpressing LuxS repressed yncC expression (Figure 4), and deleting luxS significantly increased yncC expression in MG1655 (1.3±0.1-fold). Since YncC represses ybiM, this also suggests that overexpression of LuxS can induce ybiM expression. This agrees with the previous finding that AI-2 induces ybiM expression (DeLisa et al., 2001b). Together, these results raise the possibility LuxS and its signal AI-2 may regulate yncC and ybiM through both metabolic and signaling pathways.
Overexpressing the AI-2 exporter TqsA, lsr-operon regulators LsrR and LsrK, and quorum-sensing regulator MqsR induced yncC expression. Whether these induction effects are by derepressing yncC expression via its repressor B3023 and how these effects are related to yncC negative self-regulation needs to be investigated further. For all of these tests, yncC expression levels were measured when bacterial cells were grown into the stationary phase after 6-h incubation in LB medium at 37 °C since internalization of AI-2 takes place in the stationary phase (Xavier and Bassler, 2005). Moreover, MG1655 yncC and MG1655 have the same specific growth rate during the early exponential phase, but obtain different cell densities at the late exponential stage and stationary stage (the yncC mutant enters stationary stage with a significantly lower cell density, turbidity of 600 nm of 2.7±0.2 vs 5.3±0.1 for the wild-type strain). This suggests YncC functions at the late exponential stage and stationary stage, which are consistent with the finding that the homolog of E. coli yncC in the Gram-negative bacterium Salmonella enterica serovar typhimurium is under the regulation of the stationary-phase sigma factor σS (RpoS) (Ibanez-Ruiz et al., 2000). This mimics bacterial AI-2 internalization behavior (Xavier and Bassler, 2005) and also agrees with the concept that RpoS is intimately associated with E. coli quorum sensing (DeLisa et al., 2001a), which suggests that the quorum-sensing process, stress response process and biofilm development are tightly linked.
This study reveals that the MqsR-regulated yncC gene is important for biofilm formation, and that YncC regulates E. coli biofilm formation and mucoidy via colanic acid production by repressing expression of ybiM, which encodes a putative periplasmic protein. Further characterization of B3023 and YbiM may link bacterial signaling pathways with biofilm formation. We propose yncC should be named mcbR (MqsR-controlled colanic acid and biofilm regulator), and ybiM, the first gene regulated by mcbR, should be mcbA. A conceptual model of how yncC and AI-2 influence biofilm formation is shown in Figure 6.

Conceptual model for yncC regulation of biofilm formation. → indicates induction; ⊥ indicates repression; dashed lines indicate indirect regulation and solid lines indicate direct regulation.
Accession codes
References
Andrews SC, Berks BC, McClay J, Ambler A, Quail MA, Golby P et al. (1997). A 12-cistron Escherichia coli operon (hyf) encoding a putative proton-translocating formate hydrogenlyase system. Microbiology 143: 3633–3647.
Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M et al. (2006). Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2: 2006.0008.
Bansal T, Englert D, Lee J, Hegde M, Wood TK, Jayaraman A . (2007). Differential effects of epinephrine, norepinephrine, and indole on Escherichia coli O157:H7 chemotaxis, colonization, and gene expression. Infect Immun 75: 4597–4607.
Beloin C, Valle J, Latour-Lambert P, Faure P, Kzreminski M, Balestrino D et al. (2004). Global impact of mature biofilm lifestyle on Escherichia coli K-12 gene expression. Mol Microbiol 51: 659–674.
Benjamini Y, Hochberg Y . (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Royal Stat Soc B 57: 289–300.
Blattner FR, Plunkett 3rd G, Bloch CA, Perna NT, Burland V, Riley M et al. (1997). The complete genome sequence of Escherichia coli K-12. Science 277: 1453–1474.
Camilli A, Bassler BL . (2006). Bacterial small-molecule signaling pathways. Science 311: 1113–1116.
Cherepanov PP, Wackernagel W . (1995). Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158: 9–14.
Claret L, Hughes C . (2002). Interaction of the atypical prokaryotic transcription activator FlhD2C2 with early promoters of the flagellar gene hierarchy. J Mol Biol 321: 185–199.
Clarke MB, Sperandio V . (2005). Transcriptional regulation of flhDC by QseBC and sigma (FliA) in enterohaemorrhagic Escherichia coli. Mol Microbiol 57: 1734–1749.
Da Re S, Ghigo JM . (2006). A CsgD-independent pathway for cellulose production and biofilm formation in Escherichia coli. J Bacteriol 188: 3073–3087.
Danese PN, Pratt LA, Kolter R . (2000). Exopolysaccharide production is required for development of Escherichia coli K-12 biofilm architecture. J Bacteriol 182: 3593–3596.
Danese PN, Silhavy TJ . (1998). CpxP, a stress-combative member of the Cpx regulon. J Bacteriol 180: 831–839.
Datsenko KA, Wanner BL . (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97: 6640–6645.
DeLisa MP, Valdes JJ, Bentley WE . (2001a). Mapping stress-induced changes in autoinducer AI-2 production in chemostat-cultivated Escherichia coli K-12. J Bacteriol 183: 2918–2928.
DeLisa MP, Wu CF, Wang L, Valdes JJ, Bentley WE . (2001b). DNA microarray-based identification of genes controlled by autoinducer 2-stimulated quorum sensing in Escherichia coli. J Bacteriol 183: 5239–5247.
Domka J, Lee J, Bansal T, Wood TK . (2007). Temporal gene-expression in Escherichia coli K-12 biofilms. Environ Microbiol 9: 332–346.
Domka J, Lee J, Wood TK . (2006). YliH (BssR) and YceP (BssS) regulate Escherichia coli K-12 biofilm formation by influencing cell signaling. Appl Environ Microbiol 72: 2449–2459.
Edgar R, Domrachev M, Lash AE . (2002). Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30: 207–210.
Ferrieres L, Aslam SN, Cooper RM, Clarke DJ . (2007). The yjbEFGH locus in Escherichia coli K-12 is an operon encoding proteins involved in exopolysaccharide production. Microbiology 153: 1070–1080.
Forde A, Fitzgerald GF . (2003). Molecular organization of exopolysaccharide (EPS) encoding genes on the lactococcal bacteriophage adsorption blocking plasmid, pCI658. Plasmid 49: 130–142.
Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A . (2003). ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res 31: 3784–3788.
Genevaux P, Wawrzynow A, Zylicz M, Georgopoulos C, Kelley WL . (2001). DjlA is a third DnaK co-chaperone of Escherichia coli, and DjlA-mediated induction of colanic acid capsule requires DjlA-DnaK interaction. J Biol Chem 276: 7906–7912.
González Barrios AF, Zuo R, Hashimoto Y, Yang L, Bentley WE, Wood TK . (2006). Autoinducer 2 controls biofilm formation in Escherichia coli through a novel motility quorum-sensing regulator (MqsR, B3022). J Bacteriol 188: 305–316.
Gordon JJ, Towsey MW, Hogan JM, Mathews SA, Timms P . (2006). Improved prediction of bacterial transcription start sites. Bioinformatics 22: 142–148.
Gottesman S . (2004). The small RNA regulators of Escherichia coli: roles and mechanisms. Annu Rev Microbiol 58: 303–328.
Hancock V, Klemm P . (2007). Global gene expression profiling of asymptomatic bacteriuria Escherichia coli during biofilm growth in human urine. Infect Immun 75: 966–976.
Hanna A, Berg M, Stout V, Razatos A . (2003). Role of capsular colanic acid in adhesion of uropathogenic Escherichia coli. Appl Environ Microbiol 69: 4474–4481.
Herzberg M, Kaye IK, Peti W, Wood TK . (2006). YdgG (TqsA) controls biofilm formation in Escherichia coli K-12 through autoinducer 2 transport. J Bacteriol 188: 587–598.
Ho TD, Waldor MK . (2007). Enterohemorrhagic Escherichia coli O157:H7 gal mutants are sensitive to bacteriophage P1 and defective in intestinal colonization. Infect Immun 75: 1661–1666.
Ibanez-Ruiz M, Robbe-Saule V, Hermant D, Labrude S, Norel F . (2000). Identification of RpoS (σS)-regulated genes in Salmonella enterica serovar typhimurium. J Bacteriol 182: 5749–5756.
Jackson DW, Simecka JW, Romeo T . (2002). Catabolite repression of Escherichia coli biofilm formation. J Bacteriol 184: 3406–3410.
Jeon B, Itoh K, Misawa N, Ryu S . (2003). Effects of quorum sensing on flaA transcription and autoagglutination in Campylobacter jejuni. Microbiol Immunol 47: 833–839.
Keseler IM, Collado-Vides J, Gama-Castro S, Ingraham J, Paley S, Paulsen IT et al. (2005). EcoCyc: a comprehensive database resource for Escherichia coli. Nucleic Acids Res 33: D334–D337.
Koutsoudis MD, Tsaltas D, Minogue TD, von Bodman SB . (2006). Quorum-sensing regulation governs bacterial adhesion, biofilm development, and host colonization in Pantoea stewartii subspecies stewartii. Proc Natl Acad Sci USA 103: 5983–5988.
Kumar AS, Mody K, Jha B . (2007). Bacterial exopolysaccharides—a perception. J Basic Microbiol 47: 103–117.
Lee J, Bansal T, Jayaraman A, Bentley WE, Wood TK . (2007a). Enterohemorrhagic Escherichia coli biofilms are inhibited by 7-hydroxyindole and stimulated by isatin. Appl Environ Microbiol 73: 4100–4109.
Lee J, Jayaraman A, Wood TK . (2007b). Indole is an inter-species biofilm signal mediated by SdiA. BMC Microbiol 7: 42.
Lee J, Page R, García-Contreras R, Palermino JM, Zhang XS, Doshi O et al. (2007c). Structure and function of the Escherichia coli protein YmgB: a protein critical for biofilm formation and acid-resistance. J Mol Biol 373: 11–26.
Li J, Attila C, Wang L, Wood TK, Valdes JJ, Bentley WE . (2007). Quorum sensing in E. coli is signaled by AI-2/LsrR: effects on sRNA and biofilm architecture. J Bacteriol 189: 6011–6020.
Liu X, Matsumura P . (1994). The FlhD/FlhC complex, a transcriptional activator of the Escherichia coli flagellar class II operons. J Bacteriol 176: 7345–7351.
Lombardia E, Rovetto AJ, Arabolaza AL, Grau RR . (2006). A LuxS-dependent cell-to-cell language regulates social behavior and development in Bacillus subtilis. J Bacteriol 188: 4442–4452.
Mendrygal KE, González JE . (2000). Environmental regulation of exopolysaccharide production in Sinorhizobium meliloti. J Bacteriol 182: 599–606.
Miller WG, Leveau JH, Lindow SE . (2000). Improved gfp and inaZ broad-host-range promoter-probe vectors. Mol Plant Microbe Interact 13: 1243–1250.
Obadia B, Lacour S, Doublet P, Baubichon-Cortay H, Cozzone AJ, Grangeasse C . (2007). Influence of tyrosine-kinase Wzc activity on colanic acid production in Escherichia coli K12 cells. J Mol Biol 367: 42–53.
Ogino T, Matsubara M, Kato N, Nakamura Y, Mizuno T . (1998). An Escherichia coli protein that exhibits phosphohistidine phosphatase activity towards the HPt domain of the ArcB sensor involved in the multistep His-Asp phosphorelay. Mol Microbiol 27: 573–585.
Otto K, Silhavy TJ . (2002). Surface sensing and adhesion of Escherichia coli controlled by the Cpx-signaling pathway. Proc Natl Acad Sci USA 99: 2287–2292.
Petty NK, Evans TJ, Fineran PC, Salmond GP . (2007). Biotechnological exploitation of bacteriophage research. Trends Biotechnol 25: 7–15.
Postma PW, Lengeler JW, Jacobson GR . (1993). Phosphoenolpyruvate:carbohydrate phosphotransferase systems of bacteria. Microbiol Rev 57: 543–594.
Potrykus J, Wegrzyn G . (2004). The ypdI gene codes for a putative lipoprotein involved in the synthesis of colanic acid in Escherichia coli. FEMS Microbiol Lett 235: 265–271.
Pratt LA, Kolter R . (1998). Genetic analysis of Escherichia coli biofilm formation: roles of flagella, motility, chemotaxis and type I pili. Mol Microbiol 30: 285–293.
Puan KJ, Wang H, Dairi T, Kuzuyama T, Morita CT . (2005). fldA is an essential gene required in the 2-C-methyl-D-erythritol 4-phosphate pathway for isoprenoid biosynthesis. FEBS Lett 579: 3802–3806.
Qiu D, Eisinger VM, Rowen DW, Yu HD . (2007). Regulated proteolysis controls mucoid conversion in Pseudomonas aeruginosa. Proc Natl Acad Sci USA 104: 8107–8112.
Rader BA, Campagna SR, Semmelhack MF, Bassler BL, Guillemin K . (2007). The quorum sensing molecule AI-2 regulates motility and flagellar morphogenesis in Helicobacter pylori. J Bacteriol 189: 6109–6117.
Reed JL, Vo TD, Schilling CH, Palsson BO . (2003). An expanded genome-scale model of Escherichia coli K-12 (iJR904 GSM/GPR). Genome Biol 4: R54.1–R54.12.
Ren D, Bedzyk LA, Thomas SM, Ye RW, Wood TK . (2004a). Gene expression in Escherichia coli biofilms. Appl Microbiol Biotechnol 64: 515–524.
Ren D, Bedzyk LA, Ye RW, Thomas SM, Wood TK . (2004b). Differential gene expression shows natural brominated furanones interfere with the autoinducer-2 bacterial signaling system of Escherichia coli. Biotechnol Bioeng 88: 630–642.
Ren D, Zuo R, González Barrios AF, Bedzyk LA, Eldridge GR, Pasmore ME et al. (2005). Differential gene expression for investigation of Escherichia coli biofilm inhibition by plant extract ursolic acid. Appl Environ Microbiol 71: 4022–4034.
Rickard AH, Palmer Jr RJ, Blehert DS, Campagna SR, Semmelhack MF, Egland PG et al. (2006). Autoinducer 2: a concentration-dependent signal for mutualistic bacterial biofilm growth. Mol Microbiol 60: 1446–1456.
Rudd KE, Humphery-Smith I, Wasinger VC, Bairoch A . (1998). Low molecular weight proteins: a challenge for post-genomic research. Electrophoresis 19: 536–544.
Sambrook J, Fritsch EF, Maniatis T . (1989). Molecular Cloning, A Laboratory Manual. Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY.
Schauder S, Shokat K, Surette MG, Bassler BL . (2001). The LuxS family of bacterial autoinducers: biosynthesis of a novel quorum-sensing signal molecule. Mol Microbiol 41: 463–476.
Schembri MA, Kjaergaard K, Klemm P . (2003). Global gene expression in Escherichia coli biofilms. Mol Microbiol 48: 253–267.
Shah D, Zhang Z, Khodursky A, Kaldalu N, Kurg K, Lewis K . (2006). Persisters: a distinct physiological state of E. coli. BMC Microbiol 6: 53.
Shao H, Lamont RJ, Demuth DR . (2007). Autoinducer-2 is required for biofilm growth of Aggregatibacter (Actinobacillus) actinomycetemcomitans. Infect Immun 75: 4211–4218.
Stanley NR, Lazazzera BA . (2004). Environmental signals and regulatory pathways that influence biofilm formation. Mol Microbiol 52: 917–924.
Stevenson G, Andrianopoulos K, Hobbs M, Reeves PR . (1996). Organization of the Escherichia coli K-12 gene cluster responsible for production of the extracellular polysaccharide colanic acid. J Bacteriol 178: 4885–4893.
Taga ME, Miller ST, Bassler BL . (2003). Lsr-mediated transport and processing of AI-2 in Salmonella typhimurium. Mol Microbiol 50: 1411–1427.
Taga ME, Semmelhack JL, Bassler BL . (2001). The LuxS-dependent autoinducer AI-2 controls the expression of an ABC transporter that functions in AI-2 uptake in Salmonella typhimurium. Mol Microbiol 42: 777–793.
Tamimi Y, Lines M, Coca-Prados M, Walter MA . (2004). Identification of target genes regulated by FOXC1 using nickel agarose-based chromatin enrichment. Invest Ophthalmol Vis Sci 45: 3904–3913.
Van Houdt R, Michiels CW . (2005). Role of bacterial cell surface structures in Escherichia coli biofilm formation. Res Microbiol 156: 626–633.
Wang L, Li J, March JC, Valdes JJ, Bentley WE . (2005). luxS-dependent gene regulation in Escherichia coli K-12 revealed by genomic expression profiling. J Bacteriol 187: 8350–8360.
Wassarman KM, Storz G . (2000). 6S RNA regulates E. coli RNA polymerase activity. Cell 101: 613–623.
Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V et al. (2006). Database resources of the national center for biotechnology information. Nucleic Acids Res 34: D173–D180.
Wiame E, Delpierre G, Collard F, Van Schaftingen E . (2002). Identification of a pathway for the utilization of the Amadori product fructoselysine in Escherichia coli. J Biol Chem 277: 42523–42529.
Xavier KB, Bassler BL . (2003). LuxS quorum sensing: more than just a numbers game. Curr Opin Microbiol 6: 191–197.
Xavier KB, Bassler BL . (2005). Regulation of uptake and processing of the quorum-sensing autoinducer AI-2 in Escherichia coli. J Bacteriol 187: 238–248.
Zhang XS, García-Contreras R, Wood TK . (2007). YcfR (BhsA) influences Escherichia coli biofilm formation through stress response and surface hydrophobicity. J Bacteriol 189: 3051–3062.
Zinkewich-Peotti K, Fraser JM . (1988). New locus for exopolysaccharide overproduction in Escherichia coli K-12. J Bacteriol 170: 1405–1407.
Acknowledgements
This research was supported by the NIH (EB003872-01A1). We thank Proffesor Steven Lindow for providing plasmid pPROBE-gfp[tagless] and the National of Institute of Genetics for the KEIO and ASKA clones.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhang, XS., García-Contreras, R. & Wood, T. Escherichia coli transcription factor YncC (McbR) regulates colanic acid and biofilm formation by repressing expression of periplasmic protein YbiM (McbA). ISME J 2, 615–631 (2008). https://doi.org/10.1038/ismej.2008.24
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2008.24
Keywords
This article is cited by
-
Functioning of a tripartite lignocellulolytic microbial consortium cultivated under two shaking conditions: a metatranscriptomic study
Biotechnology for Biofuels and Bioproducts (2023)
-
The bile salt glycocholate induces global changes in gene and protein expression and activates virulence in enterotoxigenic Escherichia coli
Scientific Reports (2019)
-
Metabolome and transcriptome-wide effects of the carbon storage regulator A in enteropathogenic Escherichia coli
Scientific Reports (2019)
-
Characterization of the GntR family regulator HpaR1 of the crucifer black rot pathogen Xanthomonas campestris pathovar campestris
Scientific Reports (2016)
-
Characterization of self-generated variants in Pseudoalteromonas lipolytica biofilm with increased antifouling activities
Applied Microbiology and Biotechnology (2015)
