
Abstract
Objective:
Studies have indicated that weight regain following weight loss predisposes obese individuals to metabolic disorders; however, the molecular mechanism of this potential adverse effect of weight regain is not fully understood. Here we investigated global transcriptome changes and the immune response in mouse white adipose tissue caused by weight regain.
Design:
We established a diet switch protocol to compare the effects of weight regain with those of weight gain without precedent weight loss, weight loss maintenance and chow diet. We conducted a time course analysis of global transcriptome changes in gonadal white adipose tissue (gWAT) during the weight fluctuation. Co-expression network analysis was used to identify functional modules associated with the weigh regain phenotype. Immune cell populations in gWAT were characterized by flow-cytometric immunophenotyping. Metabolic phenotypes were monitored by histological analysis of adipose tissue and liver, and blood-chemistry and body weight/composition analyses.
Results:
In total, 952 genes were differentially expressed in the gWAT in the weight regain vs the weight gain group. Upregulated genes were associated with immune response and leukocyte activation. Co-expression network analysis showed that genes involved in major histocompatibility complex I and II-mediated antigen presentation and T-cell activation function were upregulated. Consistent with the transcriptome analysis results, flow cytometry demonstrated significant increases in subsets of T cells and proinflammatory M1 macrophages in the gWAT in the weight regain as compared to the weight gain group. In addition, upregulation of adaptive immune responses was associated with high incidence of adipocyte death and upregulation of high mobility group box 1, a well-known component of damage-associated molecular patterns.
Conclusions:
Our global transcriptome analysis identified weight regain-induced activation of adaptive immune responses in mouse white adipose tissue. Results suggest that activation of adipocyte death-associated adaptive immunity in adipose tissue may contribute to unfavorable metabolic effects of weight regain following weight loss.
Similar content being viewed by others


Main
The increasing prevalence of obesity is associated with high incidences of diabetes and chronic metabolic diseases. In general, weight loss in obese people is beneficial for preventing and treating obesity-related metabolic disease; however, long-term weight loss maintenance is difficult,1 partly because of the compensatory positive energy balance after calorie restriction through an increase in appetite and the reduction in energy consumption.2, 3, 4 It has been estimated that up to 80% of people regain a significant portion of the lost weight within 1 year.5 If weight regain events following weight loss occur repeatedly, it is referred to as weight cycling, also known as yo-yo dieting.6 Importantly, weight cycling appears to have adverse effects that predispose obese individuals to metabolic disease.7 For instance, while weight loss decreases adipose tissue inflammation and improves insulin sensitivity, weight cycling promotes fat gain, enhances adipose tissue inflammation and impairs systemic glucose tolerance.8, 9, 10 Moreover, weight cycling increases the risk of morbidity and mortality in both humans and rodent models.7 While the detrimental effects of weight regain are recognized, the mechanisms of metabolic dysfunction induced by weight regain are not fully understood.2
Obesity-induced chronic inflammation is a key component in the pathogenesis of insulin resistance and metabolic syndrome.2 Diet-induced obesity mouse models and clinical studies have suggested that hypertrophic adipose tissue in obese individuals accumulates proinflammatory macrophages11 that can contribute to inflammation and insulin resistance at the systemic level.12, 13 In addition to innate immunity, adaptive immunity is involved in adipose tissue inflammation. For example, adipose tissue in obese mice has less CD4+ regulatory T cells and T helper 2 cells than that in healthy mice.8, 9 Moreover, obesity promotes the entry of B cells, CD4+ T helper 1 cells, and CD8+ cytotoxic T cells into adipose tissue.10, 11, 12 Thus, both innate and adaptive immunity contribute to regulating obesity-induced inflammation.13 Although the mechanisms by which obese adipose tissue activates proinflammatory activation is unclear, a growing body of evidence supports that adipocyte death might be a major initiator of adipose tissue inflammation and pathologic remodeling.14 Dying/dead adipocytes are characteristically surrounded by macrophages14 in so-called crown-like structures (CLSs). The frequency of CLSs increases with pathologic expansion of adipose tissue. Dying/dead adipocytes have been investigated as a source of signals for inflammation and adipose tissue remodeling because cell death generates signals activating innate and adaptive immune responses in various contexts.
In this study, to identify molecular and cellular mechanisms involved in the adverse effects of weight regain, we examined global transcriptome changes and immune responses in white adipose tissue (WAT) during the time course of weight fluctuations using ribonucleic acid (RNA) sequencing (RNAseq). First, we established a diet switch protocol to compare a weight regain (WRG) group with a weight gain (WG) group that did not undergo prior weight loss. Histological analysis of adipose tissue and liver, and blood-chemistry and body weight/composition analyses indicated a metabolically unhealthy, obese phenotype in WRG as compared to WG. Global transcriptome profiling of gonadal (g)WAT and co-expression network analysis revealed upregulation of the immune response and leukocyte activation in WRG mice. Consistent with the RNAseq data, fluorescence-activated cell sorting (FACS) analysis demonstrated increases in M1 macrophages and T cells in WRG. Finally, WRG manifested a high incidence of adipocyte cell death and an increase in circulating HMGB1, suggesting that adipocyte death might contribute to activation of the innate and adaptive immune responses that aggravate the weight regain-induced metabolic phenotype.
Materials and methods
Experimental design and laboratory animals
Four-week-old male C57BL6/N mice were purchased from Central Lab. Animal Inc. (Seoul, Republic of Korea). The animals were housed at 24±2 °C under a 12 h light/dark cycle. They were fed a chow diet (NIH-31; Ziegler Bros., Inc., PA, USA) ad libitum, along with tap water. After 1 week of acclimatization, each mouse was randomly assigned to one of four groups (n=10 per condition), according to its weight measurement (Figure 1a). High-fat diet (HFD; 60% kcal% fat, #D12492; Research Diets, NJ, USA) was used. This experimental protocol was carried out according to the Guide for Animal Experiments (edited by the Korean Academy of Medical Sciences), and was approved by the Institutional Animal Care and Use Committee (IACUC) of Seoul National University (approval number: SNU-140205-2-1). All samples were blinded to the scientists prior to analysis.

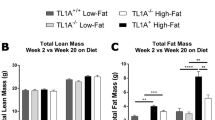
Effects of weight regain on body and tissue weight, and blood chemistry. (a) Study design for weight regain (open bar: chow diet feeding, Shaded bar: high fat diet feeding) (b) Body weight for the duration of the study. (□—control group (CTL), □—weight regain group (WRG), ▪—weight gain group (WG), △—Weight loss maintenance group (WLM)). (c) Liver weight. (d) gonadal adipose tissue (gWAT) weight. (f) visceral fat volume (e) Total fat volume. (g) Adipocyte size analysis (h) Liver autopsy image at 16 weeks on a diet. (i) Representative images of liver sections stained with hematoxylin and eosin (H&E) and Oil Red O at 16 weeks of a diet-challenge (× 200 magnification, scale bar size=100 μm). (j) Hepatic triglyceride concentration (k) Fasting glucose and (l) total cholesterol concentration. Data are presented as the mean±s.e.m.; n=10/group. *P<0.05. (a) Significantly different from the control group at the same time point. (b) Significantly different from the weight cycling group at the same time point.
Blood chemistry
Blood glucose and total cholesterol were measured in 20 μl serum samples with a Hitachi 7180 biochemistry autoanalyzer (Hitachi Ltd., Tokyo, Japan). To quantify hepatic triglyceride accumulation in the liver, the Triglyceride Quantification Colorimetric/Fluorometric Kit (K622-100; BioVision, CA, USA) was used according to the manufacturer’s instructions.
Histopathology
Each organ was weighed and fixed with 4% paraformaldehyde (Biosesang, Republic of Korea) at room temperature overnight, processed for paraffin sectioning at 4 μm thickness, and stained with hematoxylin and eosin according to standard procedures. For lipid detection in liver tissue, sections were washed once with 50% isopropyl alcohol and then stained with Oil Red O. Tissue sections were analyzed under a microscope (Olympus Co. Japan) equipped with a digital camera (DP71, Olympus). Adipocyte size was quantified from images using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Isolation of the stromal vascular fraction and FACS analysis
The stromal vascular fraction was obtained from dissociated gonadal white adipose tissue (gWAT) of mice as previously described.15 Antibodies targeting the following proteins were used for flow-cytometric analysis: CD206 (MMR; C068C2; BioLegend), CD64 (FcγRI; X54-5/7.1; BioLegend), CD45 (30-F11; BioLegend), CD11c (N418; BioLegend), CD11b (M1/70; BioLegend), I-A/I-E (Major histocompatibility complex (MHC) II; M5/114.15.2; BioLegend), F4/80 (A3-1; Serotec, NC, USA), NK1.1 (PK136; BioLegend), Siglec-F (E50-2440; BD Horizon, CA, USA), CD4 (GK1.5; BD Pharmingen, CA, USA), Foxp3 (FJK-16 s; eBioscience), CD19 (6D5; BioLegend), CD3 (17A2; BioLegend). Samples were analyzed on a FACS Canto II system (BD Biosciences, San Jose, CA, USA), using FlowJo software (Tree Star, Inc., Ashland, OR, USA). The gating strategy used for analysis is explained in Supplementary Figure S1A and S1B.
Quantitative reverse-transcription PCR
Total RNA was extracted from epididymal (e) WAT tissue using the PureLink RNA Mini Kit (Invitrogen, Carlsbad, CA, USA) and was quantified using a NanoVue spectrophotometer (GE Healthcare, NJ, USA). Complementary dioxyribonucleic acid (cDNA) was synthesized using AccuPower CycleScript RT PreMix (K-2044; Bioneer, Republic of Korea), according to manufacturer’s protocol. Quantitative PCR (qPCR) was performed with SYBR Green dye on a 7500 Real Time PCR System (Applied Biosystems, UK). Target gene expression was normalized to that of the control gene, RPLP0 ribosomal protein lateral stalk subunit P0 (36B4), and presented relative to the control group.
RNAseq
Total RNA was extracted from gWAT (60–70 mg) using the PureLink RNA Mini Kit. RNA purity was checked on the basis of the A260/A280 absorbance ratio (1.6–2.3) on a NanoVue instrument (GE Healthcare, Seoul, Republic of Korea). cDNA libraries were constructed using a TruSeq RNA Sample Preparation Kit (Illumina, San Diego, CA, USA). cDNA clusters were loaded into the Illumina HiSeq 2 000 instrument (Illumina) for automatic amplification and sequencing.
Data preprocessing and pair-wise analysis
Read and base qualities were checked with FastQC v. 0.10.1. Trimmomatic v. 0.33 was used for 3′-end trimming, processed reads were mapped onto the mouse genome (mm10/GRCm38) by using TopHat v. 2.0.13, and read counts were calculated with HTSeq v. 0.6.0. Fragments Per Kilobase of exons for per Million (FPKM) was calculated to represent 22 083 individual protein-coding genes (GRCm38), using a published formula.16 Sample outliers were excluded on the basis of sample connectivity score Z.k (standardized connectivity)<–2.5 calculated with flashClust in R.17 Sample #1 of the weight maintain group (Z.k<–3.66) was eliminated for further analysis. To improve statistical power, low-read-count genes were removed in case of Count Per Million>2 in at least 3 samples.18 Finally, 13 193 expressed genes were used for further analysis. To assess differentially expressed genes (DEGs), a generalized linear model was used in the EdgeR package in R v. 3.1.2. For pairwise analysis, DEGs were defined at absolute log2 fold change>1.0 and false discovery rate (FDR) (Benjamini & Hochberg correction)<0.05. Gene set analysis (GSA) on DEGs was performed using Databases for functional Annotation, Visualization, and Integrated Discovery (DAVID) v. 6.7 for functional analysis. Top-ranked biological functions were sorted by P-value. Significantly enriched functions were defined at FDR<0.15. Raw read counts and processed data are accessible in the Gene Expression Omnibus public database (record number: GSE73345). Within and between groups the variance showed similar scale of expression.
Network analysis of the weight regain model
In total, 2219 DEGs identified with absolute log2 fold change>1.5 and FDR<0.05 at each diet-challenge phase were used for network construction. Unsigned adjacency matrix (power=12) was constructed according to the general procedure of weighted gene co-expression network analysis (WGCNA).19 Next, individual gene modules were defined, each with a minimum of 30 genes. A global gene-gene co-expression network was constructed at adjacency threshold>0.6 (no. of genes=1416). In the global network, functional modules reflecting body weight fluctuation were enriched by Kyoto encyclopedia of genes and genomes (KEGG) pathway analysis in DAVID. The network was clustered by using the Markov Cluster Algorithm.20 Network visualization and clustering were performed with Cytoscape v. 3.2.1.
Statistical analyses
Results are expressed as the mean±s.e.m. SPSS (version 14.0, IBM, NY, USA) was used for all statistical analyses. Data were analyzed using one-way analysis of variance (ANOVA), followed by a post hoc Student’s t-test provided the ANOVA result was significant. Two-way ANOVA was used to compare measurements with two different variables. A P-value<0.05 was considered statistically significant.
Results
Weight regain following weight loss increases fat gain and is associated with adverse metabolic effects
To investigate the effects of weight regain on the metabolic phenotype, mice were randomly assigned to four experimental groups: (1) a chow control (CTL) group that received a chow diet throughout the study; (2) a weight regain (WRG) group that was placed on HFD for 8 weeks followed by chow diet for 4 weeks, and then returned to HFD for 4 weeks; (3) a weight gain (WG) group that was maintained on a chow diet for 12 weeks followed by HFD for 4 weeks; (4) a weight loss maintenance (WLM) group that received 8 weeks of a HFD and 8 weeks of chow diet (Figure 1a). As shown in Figure 1a, each group was analyzed at 8, 12 and 16 weeks.
Body weight analysis indicated that WRG mice lost weight to a level similar to that in control mice by week 12 (Figure 1b). However, WRG mice gained more weight when they received HFD than the WG group that did not undergo weight loss before HFD. Similarly, weights of white adipose tissue depots and liver in the WRG group were significantly higher than in the WG group (Figures 1c–f). Adipocyte size was significantly increased in the WRG group compared to the weight gain group (Figure 1g). In addition, WRG mice accumulated lipid droplets, resembling macro- and micro-vesicular steatosis, in their livers as indicated by histology (Figures 1h and i) and triglycerol contents (Figure 1j). Moreover, fasting blood glucose and total cholesterol were significantly increased in the WRG group as compared to the WG group (Figures 1k and l). Taken together, these findings indicate that weight regain after weight loss negatively affects metabolic health as compared to weight gain.
Transcriptome profiling of gWAT of WRG mice following weight loss
To characterize transcriptional changes induced by weight regain following weight loss, transcript expression levels in the gWAT of all groups were analyzed by RNAseq. DEGs were identified and subjected to functional analysis with DAVID. After HFD challenge from weeks 5 to 13, 1905 genes were identified as DEGs between the WRG and CTL groups; these genes were enriched for cell cycle and skeletal system development functions (Supplementary Table S1). Upon chow diet from 13 to 17 weeks, 1923 genes were identified as DEGs between the WRG and CTL groups that were associated with immune response, cell cycle, and ion transport (Supplementary Table S1). After HFD challenge from weeks 17 to 21, 2100 DEGs between the WRG and CTL group were significantly enriched for cell cycle- and skeletal system development-related functions (Supplementary Table S1). In the same experimental period, 786 DEGs were identified between WG and CTL that were enriched for cell cycle and tube development functions (Supplementary Table S1). We identified 28 DEGs between the WLM and CTL groups for which no functional association was found (Supplementary Table S1). To assess the effect of WRG, we compared the WRG and WG groups upon HFD challenge from weeks 17 to 21. In total, 952 DEGs were identified, 776 of which were upregulated and were enriched for functions associated with immune cell activation (immune response, leukocyte activation, and cell activation); the downregulated DEGs did not show significant enrichment for a function (Supplementary Table S1). These results suggest that HFD challenge primarily modulates cell cycle, skeletal system development, and immune response. Furthermore, WRG may affect immune cell activation function.
Co-expression network analysis of white adipose tissue of WRG mouse model
To construct a co-expression network for the weight regain model, 2219 DEGs were selected from pairwise comparison of the groups at each of the three diet-challenge phases. In total, 12 modules were identified by using Weighted Gene Co-Expression Network Analysis (WGCNA; Supplementary Table S2). The expression patterns in 5 functional modules (1, 3, 5, 7, 8 and 10) showed a significant correlation with body weight in the WRG model (Figure 2, Supplementary Table S2). Genes in the module 1 were mainly associated with cell cycle accordingly to gene ontology (Supplementary Table S2). The module 3 was enriched for genes involved in the innate immune response on the basis of gene ontology and pathway analyses (Supplementary Table S2, Figure 2). Several Toll-like receptors (Tlr1, 2, 6, 7, and 11) and NOD-like receptor family, pyrin domain-containing 3 (Nlrp3) were found in this module. The module 5 was enriched for genes involved in cell motion function, including arachidonate 12-lipoxygenase, 12 S-type (Alox12), bone morphogenetic protein receptor type-1B (Bmpr1b), cholecystokinin (Cck), proheparin-binding EGF-like growth factor (Hbegf) and COUP transcription factor 1 (Nr2f1). The module 7 was enriched for cell activation, leukocyte migration and chemokine signaling pathway genes. The module 8 was significantly enriched for genes related to immune response (Cd4 antigen (Cd4), Cd8 antigen (Cd8a and Cd8b1), Cd40 antigen (Cd40), chemokine (C–C motif) antigen 5 (Ccl5/RANTES)) and antigen processing/presentation pathway (Class II transactivator (CIITA), MHC class I: H2-Q6, class II: H2-Aa, H2-Eb1, H2-DMa). Expression of most of the genes in the module 8 was boosted between weeks 17 and 21(Figure 2). Particularly, body weight in the WRG group was boosted and higher than that in other groups from weeks 17 to 21 (Figure 1b). These results suggest that the functions in the module 8 reflect the metabolic response to weight regain and may have role in the weight regain-induced adaptive response in gWAT.

Body weight associated functional modules in global gene co-expression network. Heatmap (mean of log2 transformed FPKM, scaled by ‘gene’) was drawn from each module which correlated with BW at WRG time points, and was enriched to KEGG pathway by using DAVID. Left: pathway, Right: P value (FDR<0.15; also see Supplementary Table S2).
Topology-based subnetwork analysis
To identify effector genes, we constructed a subnetwork using a topology-based method. We focused on the module 8 because the changes in gene expression in this module coincided with the body-weight fluctuation pattern during the three phases of WRG. In total, 145 subnetworks were predicted by the Markov Cluster Algorithm, 14 of which harbored at least one gene present in the module 8 (Figure 3, Supplementary Table S3). These subnetworks were enriched for genes related to innate immune response, T-cell properties, and MHC class II (Figure 3a). The module 8 and the subnetworks were significantly enriched for antigen processing/presentation pathway, which are the primary function activating adaptive immune cell-like T cells.21, 22 To assess the genes associated with antigen processing/presentation further, we selected adaptive immunity genes, such as those encoding MHC molecules, costimulators, and their transcription regulators (Figure 3b, Supplementary Table S4). Genes encoding several MHC class II molecules (H2-Ab1, H2-DMa, H2-DMb1, H2-Eb1, and Cd74) and the costimulatory molecule CD83 (Cd83) were present in the selected subnetworks (Figure 3, Supplementary Table S4). The expression of the selected genes generally followed the body weight fluctuation and increased after the second HFD challenge (Figure 3, Supplementary Table S4). Particularly, several key MHC class II genes (H2-Ab1, H2-Eb1, and Cd74) showed a high FPKM value (Supplementary Table S4). qPCR was used to validate the expression of these genes. Consistent with the RNAseq results, MHC class II genes were increased in the WRG group as compared to the CTL group (Supplementary Figure S1C). Expression of H2-Eb1 and Cd74 did not differ between the WG and CTL groups (Figure 3 and Supplementary Table S3). These results suggest that the expression of MHC class II genes is affected by WRG.

Weight regain responding modules and associated subnetworks. (a) 14 subnetworks (102 genes, orphans were pre-excluded at no. of genes<2) and their functions of genes in weight regain responding modules. (b) Heatmap for genes related MHC class I, II, costimulators and regulators upregulated in gWAT of WRG groups. *: components of the subnetworks. a—vs CTL group of each time points (week 8, 12, 16), b—vs WLM, c—vs WG of week 16 at FDR<0.05.
Immune cell responses in adipose tissue of WRG mice
To determine whether weight regain modulates immune cell composition in adipose tissue, the stromal vascular fraction of gWAT was analyzed by flow cytometry (Figure 4). Both the total number of macrophages and the number of M1 macrophages in particular were significantly higher in the WRG than in the WG group (Figures 4a and b; P<0.05). Consistent with the findings for the macrophage subpopulation, the numbers of dendritic cells (DCs) were higher in the WRG than in the WG group (Figure 4b). In addition, the M1-to-M2 ratio was higher in the WRG group relative to the WG group (Figure 4c; P<0.05). Thus, weight regain modulated the activation of M1 macrophages. The WRG group had higher numbers of B cells and total T cells, including CD4+ T cells, than the WG group (Figure 4d; P<0.05). In contrast, the number of regulatory T cells (Treg), which are involved in anti-inflammatory responses, did not differ between the WRG and WG groups (Figure 4d). The Treg-to-CD4+ T-cell ratio was significantly lower in the WRG than in the WG group (Figure 4e; P<0.05). These data indicate that the population levels of both innate and adaptive immune cells influence adipose tissue inflammation in WRG mice.

Flow cytometric analysis of immune cell population in gWAT of weight regain mouse model (a) Flow profiles of stromal vascular fraction cells isolated from gWAT of CTL, WRG, WG and WLM mice. (b) Quantification of macrophage subsets and dendritic cells. (CD64+CD11b+ population represents total macrophages. CD11c-positive cells (M1 macrophages), CD206-positive cells (M2 macrophages), MHC II and CD11c-positive cells (dendritic cells) (c) M1 to M2 ratio at 16 weeks of diet-challenge. (d) Quantification of the adaptive immune cell population, gated using CD3 and CD19, to identify T cells and B cells. CD3-positive gated cells were analyzed for Foxp3, CD4-Foxp3-positive cells (Regulatory T cells, Treg), and CD4-positive cells (CD4+ T cells) were analyzed. (e) Treg to CD4+ T-cell ratio at 16 weeks of diet-challenge. Data are presented as the mean±s.e.m.; n=10–12/group. *P<0.05.
Weight regain is associated with high incidence of adipocyte cell death and an increase in cytosolic HMGB1 in gWAT
To identify adipose tissue-specific genes from the selected subnetworks, we assessed their expression in multiple mouse tissues. From 14 subnetworks, we selected 20 genes that were enriched in adipose tissue using GeneAnalytics in the GeneCards database, which provides GSA results for matched tissues (Table 1). Several genes related to MHC molecules (H2-Aa, CD83 and Cd74), T-cell activation (Ccl5) and development (IKAROS family zinc finger 1 (Ikzf1), RAS guanyl-releasing protein 1 (Rasgrp1)), and autophagy (immunity-related GTPase family M members 2 (Irgm2)) belonged to the candidates (Table 1). To confirm upregulation of genes related to MHC and immune responses, we performed qPCR analysis and demonstrated that expression levels of selected genes increased in the WRG group compared to WG group (Figure 5a). Cell death is one of the major signals that can activate innate immunity. Interestingly, the WRG contained more CLSs in gWAT than the WG group (Figure 5b). Innate immunity can be activated by pathogen-associated or damage-associated molecular patterns (PAMPS/DAMPS). Thus, we examined the well-known DAMP, HMGB1, in adipocytes. Histological analysis indicated that HMGB1 was localized in the cytoplasmic fraction in the WG group, while in CTL adipose tissue, HMGB1 was localized in the nucleus mostly. Circulating HMGB1 correlates with the weight regain phenotype (Figure 5c). Thus, weight regain increased hypertrophic adipocytes and CLSs, contributing to adipose tissue inflammation.

Increase in adipocyte death and changes in immune cell markers in gWAT of weight regain mouse model. (a) Expression of immune cell markers in adipose tissue. (b) Representative images of gWAT sections stained with H&E, and immunohistochemistry with antibodies against CD11c (× 200 magnification, scale bar size=100 μm). (c) Quantification of crown-like structure (CLS) (d) immune fluorescent staining of HMGB1 in gWAT of WRG and WG mice. Data are presented as the mean±s.e.m.; n=10/group. *P<0.05.
Discussion
Nutritional interventions and calorie restriction aimed at weight loss are often accompanied by repeated bouts of weight loss and regain.6 This study aimed to investigate a weight regain model system to clarify the molecular mechanisms of weight regain-induced metabolic consequences in adipose tissue. To this end, we established a diet switch protocol to model a series of weight fluctuations, consisting of an initial 8 weeks of weight gain, 4 weeks of weight loss, and 4 weeks of weight regain. Using this model, we showed that weight regain promotes elevated weight and increased fasting blood glucose and cholesterol levels as compared to weight gain.
To provide molecular clues of the mechanisms involved in the weight regain phenotype, we performed extensive transcriptome profiling of the gWAT during the progression of weight gain, weight loss and weigh regain. Weight regain specifically upregulated genes involved in the immune response, including proinflammatory cytokines that are involved in M1 macrophage polarization in adipose tissue (for example, Ccl2, Mcp1 and Ccr5),23 and interleukin 7 (Il7) and its receptor (Il7r), which play important roles in the development of B cells and the maintenance of activated T cells.24, 25 In addition, genes upregulated in WRG vs WG were significantly enriched for adaptive immune function, particularly, T-cell activation (Cd3d, Cd3e, Cd4, Cd8a and Ccl5).
Furthermore, co-expression network analysis yielded five functional modules that showed a correlation with body weight fluctuations during the weight regain protocol. In particular, one module (module 8) showed expression profiles similar to the body weight fluctuation, and gene expression was boosted after weight regain in this module. The module 8 was enriched for genes related to immune response and antigen processing/presentation pathway. Several recent reports have proposed that the activation loop between MHC class II and CD4 T cells, particularly in adipose tissues, is modulated by diet-induced obesity.21, 22 For network topology, we selected 14 subnetworks that contained more than one module 8 gene. These subnetworks were enriched for innate immune response, T-cell activation, MHC class II, and a mature DC marker. MHC class II molecules are predominantly expressed in professional antigen-presenting cells, such as macrophages, DCs, and B cells, and are increased in diet-induced obese adipose tissue.21, 22 In addition, MHC class II has been reported as key mediator of innate, adaptive immunity and obesity in adipose tissue.21, 22, 26, 27 It would be informative to investigate whether knockout of MHC class II genes can normalize the WRG-induced adaptive response.
In addition to network analysis, we identified 20 candidate genes that were upregulated specifically in WRG as compared to WG. Upregulation in obese condition of 8 of these genes (Agt, Aif1, C1qb, Ccl5/RANTES, Cd3g, Cd74, Cd83, and Ly86) has been previously reported.8, 28 For example, Ly86/MD-1, which forms a complex with radioprotective 105 (RP105), increases in obesity-induced inflammation in a lipopolysaccharide-dependent manner, cooperating with TLR4,29 and Ly86/MD-1-deficient mice are protected from diet-induced obesity.293031
Consistent with RNAseq analysis, FACS analysis demonstrated that the adipose tissues of WRG contain a larger population of immune cells involved in innate immunity (for example, total and M1 macrophages, DCs and natural killer cells) and adaptive immunity (for example, total T cells and B cells). While adipose tissue macrophages play a central role in adipose tissue inflammation, several types of leukocytes have been involved in adipose tissue inflammation. For example, B cells have been shown to promote inflammation and regulate T-cell function during obesity38. Taken together, these data suggest that weight regain-induced distinct activation of innate and adaptive immune cells contributes to the pro-inflammatory state and unfavorable metabolic phenotype. Although the mechanisms of the distinct immune response have not been addressed in the current study, the gut microbiota may be a regulatory factor that affects adipose tissue immunity. In this regard, recent studies by Thaiss et al.32 demonstrated that an intestinal microbiome signature of obese mice persisted after dieting and could contribute to weight regain metabolic phenotypes. Further studies are required to investigate the mechanisms of adaptive and innate immunity that lead to the differential adipose tissue inflammation in WRG mice in greater detail.
Interestingly, the number of dying/dead adipocytes appearing as CLSs was higher in the adipose tissue of the WRG group. Dying adipocytes have been reported to trigger adipose tissue remodeling. Although our study did not address the characteristics of the adipocyte cell death process, it is suggested that dying/dead adipocytes could contribute to proinflammatory response and T-cell activation in the weight regain phenotype. Cytosolic HMGB1 was prominent in WRG mice, suggesting that DAMP signaling is involved in the distinct immune responses. Further investigation is needed to characterize initial immunological stimuli that cause adipocyte death and activation of adaptive immunity.
Collectively, alterations of immune cell populations and their gene expression within adipose tissue might contribute to the aggravated metabolic phenotype that occurs during weight regain. The results of this study suggested that activation of adipocyte death-associated adaptive immunity in adipose tissue might contribute to the unfavorable metabolic effects of weight regain following weight loss. Further investigation may lead to the identification of novel molecular targets for reducing the adverse effect of weight regain.
References
Greenway FL . Physiological adaptations to weight loss and factors favouring weight regain. Int J Obes 2015; 39: 1188–1196.
MacLean PS, Higgins JA, Giles ED, Sherk VD, Jackman MR . The role for adipose tissue in weight regain after weight loss. Obes Rev 2015; 16 (Suppl 1): 45–54.
Maclean PS, Bergouignan A, Cornier MA, Jackman MR . Biology's response to dieting: the impetus for weight regain. Am J Physiol Regul Integr Comp Physiol 2011; 301: R581–R600.
MacLean PS, Higgins JA, Jackman MR, Johnson GC, Fleming-Elder BK, Wyatt HR et al. Peripheral metabolic responses to prolonged weight reduction that promote rapid, efficient regain in obesity-prone rats. Am J Physiol Regul Integr Comp Physiol 2006; 290: R1577–R1588.
Kraschnewski JL, Boan J, Esposito J, Sherwood NE, Lehman EB, Kephart DK et al. Long-term weight loss maintenance in the United States. Int J Obes 2010; 34: 1644–1654.
Blackburn GL, Wilson GT, Kanders BS, Stein LJ, Lavin PT, Adler J et al. Weight cycling: the experience of human dieters. Am J Clin Nutr 1989; 49 (5 Suppl): 1105–1109.
Mehta T, Smith DL Jr., Muhammad J, Casazza K . Impact of weight cycling on risk of morbidity and mortality. Obes Rev 2014; 15: 870–881.
Anderson EK, Gutierrez DA, Kennedy A, Hasty AH . Weight cycling increases T-cell accumulation in adipose tissue and impairs systemic glucose tolerance. Diabetes 2013; 62: 3180–3188.
Barbosa-da-Silva S, Fraulob-Aquino JC, Lopes JR, Mandarim-de-Lacerda CA, Aguila MB . Weight cycling enhances adipose tissue inflammatory responses in male mice. PLoS ONE 2012; 7: e39837.
Olson MB, Kelsey SF, Bittner V, Reis SE, Reichek N, Handberg EM et al. Weight cycling and high-density lipoprotein cholesterol in women: evidence of an adverse effect: a report from the NHLBI-sponsored WISE study. Women's Ischemia Syndrome Evaluation Study Group. J Am Coll Cardiol 2000; 36: 1565–1571.
Lumeng CN, Bodzin JL, Saltiel AR . Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 2007; 117: 175–184.
Sun K, Kusminski CM, Scherer PE . Adipose tissue remodeling and obesity. J Clin Invest 2011; 121: 2094–2101.
Exley MA, Hand L, O'Shea D, Lynch L . Interplay between the immune system and adipose tissue in obesity. J Endocrinol 2014; 223: R41–R48.
Strissel KJ, Stancheva Z, Miyoshi H, Perfield JW 2nd, DeFuria J, Jick Z et al. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes 2007; 56: 2910–2918.
Kwon HJ, Kim SN, Kim YA, Lee YH . The contribution of arachidonate 15-lipoxygenase in tissue macrophages to adipose tissue remodeling. Cell Death Dis 2016; 7: e2285.
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B . Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 2008; 5: 621–628.
Horvath S (ed.) Integrated weighted correlation network analysis of mouse liver gene expression data Weighted Network Analysis-Applications in Genomics and Systems Biology. Springer Book, 2011.
Robinson MD, McCarthy DJ, Smyth GK . edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010; 26: 139–140.
Langfelder P, Horvath S . WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008; 9: 559.
Enright AJ, Van Dongen S, Ouzounis CA . An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res 2002; 30: 1575–1584.
Cho KW, Morris DL, DelProposto JL, Geletka L, Zamarron B, Martinez-Santibanez G et al. An MHC II-dependent activation loop between adipose tissue macrophages and CD4+ T cells controls obesity-induced inflammation. Cell Rep 2014; 9: 605–617.
Morris DL, Cho KW, Delproposto JL, Oatmen KE, Geletka LM, Martinez-Santibanez G et al. Adipose tissue macrophages function as antigen-presenting cells and regulate adipose tissue CD4+ T cells in mice. Diabetes 2013; 62: 2762–2772.
Morris DL, Singer K, Lumeng CN . Adipose tissue macrophages: phenotypic plasticity and diversity in lean and obese states. Curr Opin Clin Nutr Metab Care 2011; 14: 341–346.
Fry TJ, Mackall CL . The many faces of IL-7: from lymphopoiesis to peripheral T cell maintenance. J Immunol 2005; 174: 6571–6576.
Lee M, Song SJ, Choi MS, Yu R, Park T . IL-7 receptor deletion ameliorates diet-induced obesity and insulin resistance in mice. Diabetologia 2015; 58: 2361–2370.
Xiao L, Yang X, Lin Y, Li S, Jiang J, Qian S et al. Large adipocytes function as antigen-presenting cells to activate CD4 T cells via upregulating MHCII in obesity. Int J Obes (2005) 2013.
Deng T, Lyon CJ, Minze LJ, Lin J, Zou J, Liu JZ et al. Class II major histocompatibility complex plays an essential role in obesity-induced adipose inflammation. Cell Metab 2013; 17: 411–422.
Wu H, Ghosh S, Perrard XD, Feng L, Garcia GE, Perrard JL et al. T-cell accumulation and regulated on activation, normal T cell expressed and secreted upregulation in adipose tissue in obesity. Circulation 2007; 115: 1029–1038.
Watanabe Y, Nakamura T, Ishikawa S, Fujisaka S, Usui I, Tsuneyama K et al. The radioprotective 105/MD-1 complex contributes to diet-induced obesity and adipose tissue inflammation. Diabetes 2012; 61: 1199–1209.
Zhou XJ, Zhang H . Autophagy in immunity: implications in etiology of autoimmune/autoinflammatory diseases. Autophagy 2012; 8: 1286–1299.
Petkova DS, Viret C, Faure M . IRGM in autophagy and viral infections. Front Immunol 2012; 3: 426.
Thaiss CA, Itav S, Rothschild D, Meijer MT, Levy M, Moresi C et al. Persistent microbiome alterations modulate the rate of post-dieting weight regain. Nature 2016; 540: 544–551.
Acknowledgements
This research was supported by Korea Mouse Phenotyping Project (2013M3A9D5072550) and Bio & Medical Technology Development Program (2012M3A9B6055344) of the National Research Foundation (NRF) funded by the Ministry of Science and ICT (2012M3A9D1054622, 2013M3A9B6046417). This research was also supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI13C2148).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on International Journal of Obesity website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Kyung, D., Sung, H., Kim, Y. et al. Global transcriptome analysis identifies weight regain-induced activation of adaptive immune responses in white adipose tissue of mice. Int J Obes 42, 755–764 (2018). https://doi.org/10.1038/ijo.2017.297
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ijo.2017.297
