
Abstract
KIF11 mutations are known to cause autosomal dominant microcephaly-lymphedema-chorioretinopathy dysplasia syndrome, associated or not with intellectual disability. We report a father and two children presenting microcephaly, chorioretinopathy and mild intellectual disability associated with a 209-kb microdeletion at 10q23.33. This microdeletion encompasses the entire KIF11 gene. In addition to point mutations, KIF11 haploinsufficiency due to a deletion is causally associated with autosomal dominant microcephaly, chorioretinopathy and mild intellectual disability.
Similar content being viewed by others


In 2012, several different point mutations were identified in the KIF11 gene in patients from 15 different families affected by microcephaly, chorioretinopathy and lymphedema syndrome (MCLMR; MIM152950).1 Since then, other patients with KIF11 mutations have been identified, confirming the causal association between this gene and MCLMR syndrome.2–5 KIF11, which maps at 10q23.33, encodes a spindle motor protein of the kinesin family. Although KIF11 is primarily associated with MCLMR syndrome, the clinical features are heterogeneous, indicating both variable expressivity and incomplete penetrance.5 We report on the first microdeletion encompassing the entire KIF11 gene, which cosegregates in a family with true microcephaly (microcephalia vera), chorioretinopathy and mild intellectual disability.
Two siblings, a boy and his younger sister, were referred to our genetic service presenting true microcephaly (microcephalia vera) and mild intellectual disability/learning difficulty. They were the youngest children born to a nonconsanguineous couple; their older brother was clinically normal. The institutional Research Ethics Committee approved the research protocol, and written informed consent was obtained from the patients’ parents.
On physical examination at the age of 10 years, the boy presented with microcephaly (head circumference 49 cm; <1st percentile, −3.0 SD), narrow palpebral fissures and large prominent ears. He started walking without support at the age of 1 year, and speech development occurred after 3 years of age. He presented with attention deficit, hyperactivity and sleeping difficulties. Learning difficulties became apparent at school, and he could not learn to read, write or develop basic mathematical skills. Despite these limitations, at 10 years of age, he was interactive, socially disinhibited and aware of his environment and surroundings, suggesting that his cognitive impairment was mild, particularly impacting formal learning. His karyotype was normal, and testing of the FMR1 gene revealed a premutation [(CGG)57] inherited from his clinically normal mother.
His sister was examined at the age of 5 years. In addition to microcephaly (head circumference: 46 cm; <1st percentile, −3.1 SD) and speech acquisition delay, she presented bilateral strabismus. A ridge over the midline of the forehead pointed to premature closure of the metopic suture. She did not carry the FMR1 premutation.
Their father had a diagnosis of true microcephaly with no significant dysmorphic features. He was seen in our service at the age of 45 years (head circumference: 52 cm; 2nd percentile, −2.11 SD) when the family returned for the results of the genetic tests. He could not read nor write and worked as a farm laborer all his life.
Genomic DNA was extracted from peripheral blood leukocytes of the affected siblings and their parents. Chromosome microarray analysis was performed using the Cytosure ISCA 180 K v2 platform (Oxford Gene Technology) according to the manufacturer’s recommendations. Analysis was conducted as described previously.6
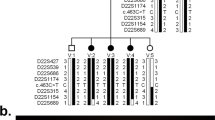
The three members of the family with microcephaly (the father and his two children) were found to carry a heterozygous microdeletion encompassing a ~209- kb segment at 10q23.33: arr[hg38] 10q23.33(92,458,987_92,667,951)x1 (ISCN 2016) (Figure 1a). This microdeletion, encompassing the entire KIF11 gene and part of the IDE (Insulin Degrading Enzyme) gene, has not been reported in the general population7 (Figure 1b). Additionally, a heterozygous duplication of a 281 kb segment mapped at 14q31.3 was detected in both children, which was inherited from their normal mother (data not shown); this variant, already reported in the general population, was considered a benign variant.

Microdeletion at 10q23.33 encompassing the KIF11 gene detected in the father and two children presenting microcephaly, mild intellectual disability and chorioretinopathy. The image shows the array comparative genomic hybridization (aCGH) profile of the deleted chromosomal segment at 10q23.33, encompassing the KIF11. The upper panel (a) shows the entire chromosome 10 copy number profile with the deletion marked by a blue vertical line at 10q23.33; below, a closer view of the deleted segment, with superposition of two array-CGH experiments with dye swap (colored regions depicting the alteration); the blue bar at the right indicates the KIF11 gene location; images adapted from Genomic Workbench software (Agilent). The lower panel (b) depicts genomic features of the 10q23.33 deleted segment according to the array-CGH mapping (arr[hg38] 10q23.33(92,458,987_92,667,951)x1 (ISCN 2016)) —the curated isoforms of the two affected genes (NCBI Ref Seq genes track), part of IDE and the entire KIF11, are shown as dark blue lines, in which the vertical bars denote exons; DGV gain and loss loci are shown as blue and red bars, respectively (images derived from the UCSC Genome Browser, freeze January 2018).
Chorioretinopathy had not been documented in any of the three patients, but after genetic testing, we conducted a detailed ophthalmological evaluation, and the father and his two children were found to present chorioretinopathy (Figure 2). The father had atrophic chorioretinal lesions with pigmented borders and discrete vitreous opacity at the lower extremity of the lesion, on the upper nasal peripheral region of the left eye (Figure 2a). Ophthalmological examination of his daughter showed bilateral strabismus, hyperopia, astigmatism and atrophic lesions of the choroid and retina in the lower temporal mid-periphery, with exposure of the sclera of both eyes (Figure 2b). The boy presented a symmetrical increase in vascular tortuosity and chorioretinal atrophy in the lower region of the retinal nerve fiber layer, adjacent to the optic disc (Figure 2c).

Color retinography of the three patients. (a) The father presents a unilateral alteration: atrophic chorioretinal lesions with pigmented borders localized on the upper nasal peripheral region of the left eye; (b) the daughter shows bilateral atrophic lesions of the choroid and retina in the lower temporal mid-periphery with exposure of the sclera; (c) the son presents symmetrical features of increase in vascular tortuosity and chorioretinal atrophy in the lower region of retinal nerve fiber layer, adjacent to the optic disc.
This is the first report on a microdeletion encompassing the entire KIF11 gene that segregates in a family with microcephaly, mild intellectual disability and chorioretinopathy, a clinical phenotype known to be caused by KIF11 point mutations. The IDE gene, partially included in the deleted segment, has been associated with susceptibility to diabetes type 2 and Alzheimer’s disease,8 both of late onset, and not documented in this family. This deletion was not reported in the general population,7 but three patients with larger deletions overlapping this segment, and presenting clinical phenotypes that included microcephaly and intellectual disability, were described in DECIPHER.9
Schlögel et al.5 reviewed the phenotypes of 87 patients carrying KIF11 point mutations from six previous studies and their own. The KIF11 point mutations were distributed throughout the gene, all of them probably resulting in loss of function. Among the mutation carriers, 91% had microcephaly, 72% eye abnormalities, 67% intellectual disability and 47% lymphedema. In addition to this variable clinical expression also observed within families, incomplete penetrance in carriers of causative mutations was reported.2 More recently, Birtel et al.10, in addition to the remarkable variability of the ocular phenotype in KIF11-related retinopathy, documented progressive retinal degeneration, thus indicating a role for KIF11 in both ocular development and maintaining retinal morphology and function. The father and his two children reported here, presented microcephaly, mild intellectual disability and a heterogeneous pattern of chorioretinopathy; progressive retinal degeneration was not apparent in the father at 45 years of age.
A biological role for kif11 in the division of stem cell populations in vivo during neural development was demonstrated in zebrafish.11 Additionally, the finding of kif11 localization in the inner segment and ciliary compartments of photoreceptor cells of the murine retina pointed to KIF11-mediated disease as representing a syndromic ciliopathy.10 Our findings show that the KIF11 haploinsufficiency due to microdeletion is associated with a clinical condition similar to that presented by patients carrying loss-of-function point mutations, further supporting the role of KIF11 alterations as strong risk factors for brain and eye developmental defects.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Ostergaard, Simpson MA, Mendola A, Vasudevan P, Connell FC, van Impel A et al. Mutations in KIF11 cause autosomal-dominant microcephaly variably associated with congenital lymphedema and chorioretinopathy. Am J Hum Genet 2012; 90: 356–362.
Jones, Ostergaard P, Moore AT, Connell FC, Williams D, Quarrell O et al. Microcephaly with or without chorioretinopathy, lymphoedema, or mental retardation (MCLMR): review of phenotype associated with KIF11 mutations. Eur J Hum Genet 2014; 22: 881–887.
Mears K, Bakall B, Harney LA, Penticoff JA, Stone EM . Autosomal dominant microcephaly associated with congenital lymphedema and chorioretinopathy due to a novel mutation in KIF11. JAMA Ophthalmol 2015; 133: 720–721.
Mirzaa, Enyedi L, Parsons G, Collins S, Medne L, Adams C et al. Congenital microcephaly and chorioretinopathy due to de novo heterozygous KIF11 mutations: five novel mutations and review of the literature. Am J Med Genet A 2014; 164A: 2879–2886.
Schlogel, Mendola A, Fastré E, Vasudevan P, Devriendt K, de Ravel TJ et al. No evidence of locus heterogeneity in familial microcephaly with or without chorioretinopathy, lymphedema, or mental retardation syndrome. Orphanet J Rare Dis 2015; 10: 52.
Krepischi, Maschietto M, Ferreira EN, Silva AG, Costa SS, da Cunha IW et al. Genomic imbalances pinpoint potential oncogenes and tumor suppressors in Wilms tumors. Mol Cytogenet 2016; 9: 20.
MacDonald JR, Ziman R, Yuen RK, Feuk L, Scherer SW . The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res 2014; 42: D986–D992.
Pivovarova O, Hohn A, Grune T, Pfeiffer AF, Rudovich N . Insulin-degrading enzyme: new therapeutic target for diabetes and Alzheimer's disease? Ann Med 2016; 48: 614–624.
Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D . DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet 2009; 84: 524–533.
Birtel J, Gliem M, Mangold E, Tebbe L, Spier I, Müller PL et al. Novel insights into the phenotypical spectrum of KIF11-associated retinopathy, including a new form of retinal ciliopathy. Invest Ophthalmol Vis Sci 2017; 58: 3950–3959.
Johnson K, Moriarty C, Tania N, Ortman A, DiPietrantonio K, Edens B et al. Kif11 dependent cell cycle progression in radial glial cells is required for proper neurogenesis in the zebrafish neural tube. Dev Biol 2014; 387: 73–92.
Data Citations
Krepischi, Ana CV HGV Database http://dx.doi.org/10.6084/m9.figshare.hgv.1758 (2018)
Acknowledgements
We are grateful to the patients’ family for participating in the study. This work was funded by FAPESP (grant CEPID - Human Genome and Stem Cell Research Center 2013/08028-1) and CNPq (PIBIC fellowship).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Malvezzi, J., H Magalhaes, I., S Costa, S. et al. KIF11 microdeletion is associated with microcephaly, chorioretinopathy and intellectual disability. Hum Genome Var 5, 18010 (2018). https://doi.org/10.1038/hgv.2018.10
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2018.10
This article is cited by
-
Burden of Rare Copy Number Variants in Microcephaly: A Brazilian Cohort of 185 Microcephalic Patients and Review of the Literature
Journal of Autism and Developmental Disorders (2024)
-
An interphase pool of KIF11 localizes at the basal bodies of primary cilia and a reduction in KIF11 expression alters cilia dynamics
Scientific Reports (2020)
