
Abstract
The mechanism underlying the vulnerability to developing schizophrenia (SCZ) during adolescence remains elusive. Hypofunction of N-methyl-d-aspartate receptors (NMDARs) has been implicated in the pathophysiology of SCZ. During development, the composition of synaptic NMDARs dramatically changes from NR2B-containing NMDARs to NR2A-containing NMDARs through the phosphorylation of NR2B S1480 or Y1472 by CDK5, CSNK2A1, and EphB2, which plays a pivotal role in the maturation of neural circuits. We hypothesized that the dysregulation of developmental change in NMDARs could be involved in the onset of SCZ. Using next-generation sequencing, we re-sequenced all the coding regions and splice sites of CDK5, CSNK2A1, and EphB2 in 474 patients with SCZ and 475 healthy controls. Variants on the database for human control subjects of Japanese origin were removed and all the nonsynonymous and nonsense variants were validated using Sanger sequencing. Four novel variants in CDK5 were observed in patients with SCZ but were not observed in controls. The total number of variants, however, was not significantly different between the SCZ and control groups (P=0.062). In silico analyses predicted P271T to be damaging. Further genetic research using a larger sample is required to examine whether CDK5 is involved in the pathophysiology of SCZ.
Similar content being viewed by others
Introduction
The onset of schizophrenia (SCZ) usually occurs during early adolescence. SCZ is a neurodevelopmental disorder with a strong genetic component and is highly related to disturbances in brain/circuit maturation.1–3 However, the molecular mechanisms underlying the vulnerability for SCZ during early adolescence remain elusive.
Accumulating evidence has demonstrated that hypofunction of N-methyl-d-aspartate receptors (NMDARs) is involved in the pathophysiology of SCZ.4–11 This hypothesis was initially based on clinical findings that the uncompetitive NMDAR antagonists, phencyclidine (PCP) and ketamine, induced psychotic features resembling symptoms seen in patients with SCZ. These features included both positive and negative symptoms, as well as cognitive dysfunction in healthy volunteers.5 In addition, PCP exacerbated these symptoms in patients with chronic SCZ.12 Further, GRIN2A, which encodes the NR2A subunit of NMDARs, has been consistently reported to be associated with SCZ 9–11 and has reached genome-wide significance in the largest genome-wide association study (GWAS) performed by the Psychiatric Genomics Consortium.13
A large exome-sequencing study using 617 trios also revealed that patients with sporadic SCZ harbored de novo mutations in genes related to NMDARs and in the activity-regulated cytoskeleton-associated protein (ARC) complex, including GRIN2A and DLG.14 Purcell et al. showed additional evidence of NMDAR hypofunction by identifying rare, disruptive mutations in NMDAR and ARC gene sets through exome-sequencing of 2536 patients with SCZ.15
NMDARs play a crucial role in synaptic plasticity, neuronal development, learning, and memory.16,17 Each NMDAR is a tetramer composed of two NR1 subunits and two NR2 or NR3 subunits.18 The number, localization and subunit composition of NMDARs are strictly regulated in an age-dependent manner so that they can exert their function precisely.16,18 The NR2 subtype, that is, NR2A-NR2D, defines the pharmacological and biophysical properties of the NMDAR.16,19,20 NR2A-containing NMDARs are considered to be more neuroprotective than NR2B-containing NMDARs depending on their Ca2+ permeability.21,22
During development, the composition of NMDARs dramatically changes from those that contain NR2B subunits to those that contain NR2A.23,24 This NMDAR subunit switch (NMDAR switch) plays an important role in the maturation of neural circuits, especially in the GABAergic inhibitory circuit in the prefrontal cortex, in a synaptic activity-dependent manner.16,18,25
Recent evidence also revealed that the NMDAR subunit switch is mediated by the endocytosis of NR2B, which is induced by phosphorylation of NR2B S1480 or Y1472 during early development.26 These phenomena are induced by cyclin-dependent kinase 5 (CDK5), casein kinase 2 (CK2), and erythropoietin-producing hepatocellular carcinoma group B (EphB2), whose activities are also regulated in an age-dependent manner.26–30
The effect of PCP on NMDARs differs according to the developmental stage and depends on the NR2 subunit composition.31,32 Postmortem brain studies in SCZ patients have shown a reduction in the mRNA expression of GRIN2A, which encodes NR2A, on parvalbumin-positive interneurons in the prefrontal cortex, and an increase in the mRNA expression of GRIN2B, which encodes NR2B, in the dorsomedial thalamus, temporal cortex, and hippocampus.6,33–35 A decrease in CK2 expression and dysregulation of CDK5-P35/P25 has been reported.36–38 CDK5 has been shown to phosphorylate the dopamine D2 receptor, which is involved in the pathophysiology of SCZ.39 In addition, 7 microRNAs that commonly regulate the expression of CDK5 were reported to be potential peripheral biomarkers for SCZ.40 Aripiprazole increased the ratio of NR2A/NR2B in rats, while risperidone increased the activity of CK2.38,41 Furthermore, EphB2 activates kalirin-7, leading to activity-dependent synaptic formation; knockdown mice for this receptor showed SCZ-like behavioral abnormalities after the period of adolescence.42
GWAS have successfully provided evidence that the common variants in genes related to synaptic function contribute to disease risk in SCZ. However, the problem of missing heritability has emerged because only 23% of the heritability can be explained by current GWAS findings.43 Identifying rare variants with a strong effect is another alternative strategy to reveal the genetic architecture of SCZ.43,44
In the current study, we hypothesized that the dysfunction of adolescence-related neurodevelopmental events, especially the NMDAR subunit switch, which is regulated by phosphorylation of NR2B, S1480, or Y1472, might contribute to the onset of SCZ. Thus, we re-sequenced CDK5 (BC005115), CSNK2A1 (BC050036), and EphB2 (BC067861) to examine the genetic contribution of these genes, focusing on rare variants to examine the pathophysiology of SCZ.
Materials and methods
Participants
A total of 474 patients with SCZ (234 males and 240 females, age 43.12±15.14 years) and 475 control subjects (224 males and 251 females, age 41.54±12.43 years) who were unrelated were analyzed in the current study. There were no significant differences in sex and age between the two groups (Student’s t-test). Inpatients and outpatients of the Department of Neuropsychiatry, University of Tokyo Hospital, and related hospitals and daycare facilities located around Tokyo were recruited for this study. Two or more senior psychiatrists diagnosed the patients through interviews and clinical records using the Diagnostic and Statistical Manual of Mental Disorders DSM-IV criteria (American Psychiatric Association). Patients with a history of drug addiction and alcohol abuse/dependence were excluded. Using the Mini-International Neuropsychiatric Interview45 or unstructured interviews, control subjects with current or lifetime mental illness, alcohol abuse/dependence, or a family history of any psychiatric disorder were also excluded. All participants were ethnically Japanese. The study was performed in accordance with the Declaration of Helsinki. The Research Ethics Committee of the Faculty of Medicine, University of Tokyo approved this study (Approval No. G0639-(33)). Written informed consent was obtained from all participants after they were provided an explanation of the study.
Re-sequencing
Genomic DNA was isolated from leukocytes derived from whole blood using a DNA blood kit (QIAGEN Ltd., Hilden, Germany), as previously described.46 A total of 250 ng of genomic double-stranded DNA was used in the target re-sequencing experiment. Target re-sequencing of all the coding regions and the splice sites (±5 base pairs (bp)) of CDK5, CSNK2A1, and EphB2 was performed to identify missense/nonsense variants and small insertions/deletions using a uniquely customized design TruSeq Custom Amplicon (Illumina, San Diego, CA, USA) with MiSeq (Illumina, California, USA) according to the manufacturer’s instructions.47 Human CDK5, CSNK2A1, and EphB2 are located on chromosomes 7q36, 20q13, and 1p36.12, respectively. Briefly, 250 ng of total double-stranded DNA was used. Amplicons were designed using Design Studio (http://designstudio.illumina.com/), and 59 amplicons were available. After hybridization, ligation, and amplification, the samples were applied to the sequencer. Raw FASTQ files from the sequencer were retrieved, and short reads were aligned to the hg19 reference using the Burrows-Wheeler Aligner software (http://bio-bwa.sourceforge.net/).48 Picard tools were used to remove duplicates. Subsequently, SAMtools (http://samtools.sourceforge.net/) was used to convert to BAM files and for variant calling.49 GATK was used for base quality score recalibration and was also used to detect SNV and small indels. VCF files were ultimately obtained, and variants were extracted. Using IGV, we visualized the next-generation sequencing data. Sanger sequencing was performed to sequence the genomic region when no amplicon was available (CDK5 exon 1 (Chr7: 150754894–150754935), exon 4 (Chr7: 150753818–150753888), exon 5 (Chr7: 150753662–150753728), CSNK2A1 exon 4 (Chr20: 485757–485878), EphB2 exon 1 (Chr1: 23037476–23037541)) and was used to validate all genetic variants using a 3130XL Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) after removal of the variants detected in a healthy database cohort of Japanese origin (Human Genetic Variation Database: http://www.genome.med.kyoto-u.ac.jp/SnpDB/index.html; HGVD). The primers were designed using Primer 3 software (http://bioinfo.ut.ee/primer3-0.4.0/primer3/). Primer pairs used in Sanger sequencing are shown in Supplementary Table S1.
In silico analysis
We predicted the functional effect of the variants that were detected using the database for nonsynonymous single nucleotide polymorphisms’ functional predictions (dbNSFP; https://sites.google.com/site/jpopgen/dbNSFP), an integrated functional analysis database of SIFT, Polyphen2, LRT, Mutation Taster, and FATHMM.50
Statistical analysis
Student’s t-test was used to compare the ages and sexes of participants in the SCZ and control groups. Fisher’s exact test was used to examine whether the number of the variants not reported in the HGVD was significantly overrepresented in patients with SCZ compared with controls.
Results
Variants observed in CDK5, CSNK2A1, and EphB2
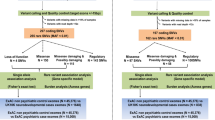
We performed target re-sequencing of CDK5, CSNK2A1, and EphB2, genes that are shown to regulate the NMDAR switch via phosphorylation of NR2B Y1472 or S1480, in 474 patients with SCZ and 475 controls. The flowchart of the analysis is shown in Figure 1. A total of 8 patients and 13 controls were excluded due to low DNA quality. All of the variants that were identified (Table 1) were novel single-base heterozygous substitutions.

Flow diagram of processes used to detect single nucleotide variations (SNVs) and insertions and deletions (indels) in N-methyl-d-aspartate (NMDA) receptor switching regulator genes in patients with schizophrenia (SCZ) and in controls. Target re-sequencing of entire coding regions and splice sites (±5 base pairs) were performed. All detected nonsynonymous variants and regions with low coverage (<20), with no available designed amplicons were examined by Sanger sequencing. After sample quality control, 8 patients and 13 controls were excluded due to low DNA quality. Three nonsynonymous and one splice region variant in CDK5 were observed in patients with SCZ but not in controls. Nonsynonymous variants of CSNK2A1 and EphB2 were identified in both patients with SCZ and controls.
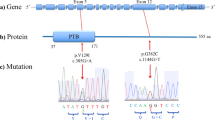
In CDK5, three nonsynonymous variants (i.e., R200Q in exon 9, P253S in exon 11, P271T in exon 12) and one splice region variant (i.e., Chr7:150751186, C to A) were observed in patients with SCZ but not in controls (Figures 2a–d).

The electropherogram traces of variants confirmed by Sanger sequencing in CDK5. The traces include four novel variants located in the exons of CDK5 (a–c) and one variant in the splice region (d). The locations, nucleotide changes, and consequent amino acid changes are described in the subfigures.
In CNSK2A1, one nonsynonymous variant (M339I) in exon 13 was observed in patients with SCZ, and two nonsynonymous variants (M339I in exon 13 and R134Q in exon 7) were observed in controls after removing the variants that were observed in the healthy cohort database.
In EphB2, we observed five nonsynonymous variants (R80H in exon 3, R229S in exon 3, V268I in exon 3, R569W in exon 9, and G734V in exon 12) and four nonsynonymous variants (N133S in exon 3, V190M in exon 3, V465M in exon 6, and R569M in exon 9) in six patients with SCZ and four controls. R569W was observed in a patient with SCZ and in a control.
The total number of variants in each gene was not significantly different between the two groups (CDK5: P=0.062, CSNK2A1: P=0.624, EphB2: P=1.000).
In silico analysis
Next, we performed a functional prediction of the variants we observed in this study using dbNSFP. In this analysis, P271T in CDK5 in patients with SCZ was predicted to be highly damaging (Table 1). Furthermore, M339I, R134Q in CSNK2A1, G734V, and V190M in EphB2 were also predicted to be highly damaging.
Discussion
To examine whether the developmental NMDAR subunit switch was genetically involved in the pathophysiology of SCZ, we performed target re-sequencing of CDK5, CSNK2A1, and EphB2, that regulate developmental NMDAR subunit switch via phosphorylation of NR2B Y1472 or S1480, using samples from 474 patients with SCZ and 475 controls. This is the first study focusing on the genetic relationship between the developmental change in NMDAR subunit switching and SCZ, although we failed to provide evidence for an NMDAR switch in the current study.
In CDK5, we observed four novel variants in patients with SCZ but not in controls, although the total number of variants detected was not significantly different between the two groups (P=0.062). The crystal structure of the cAMP-dependent protein kinase, including CDK5, has been shown to have an N-terminal domain (N lobe) that is composed of mainly ß-sheets, a helical C-terminal domain (C lobe), and a deep ATP-binding cleft between the two lobes.51 According to InterPro (https://www.ebi.ac.uk/interpro/), a database for predicting protein function, P271 is located at the helical C-terminus in CDK5 and belongs to the protein kinase domain that is crucial for its serine/threonine protein kinase activity. The tyrosine (Y1472) within the YEKL motif of NR2B was shown to be phosphorylated by the CDK5, serine/threonine protein kinase. Phosphorylation of this site inhibited the binding of AP-2 to YEKL motif, which is necessary for NR2B endocytosis, and promoted synaptic surface expression of NR2B-containing NMDARs instead of reducing the surface expression of NR2A-containing NMDARs.27 These findings raise the possibility that the P271T found in this study may affect the regulation of the developmental NMDARs switch. Another possibility is that P271T may disrupt the function of CDK5 by interrupting the formation of the CDK5–p25 complex, as the α-helix is a critical structure in the interaction between CDK5 and p25, a co-activator of CDK5.51–53 Furthermore, the C-terminus of the helix is a well-conserved region among spices51,54 and may lead to a highly damaging effect on its function.
In a previous large exome study of 2536 patients with SCZ and 2543 controls (http://research.mssm.edu/statgen/sweden/),15 a rare, nonsynonymous variant, R50W, in CDK5 was detected in patients with SCZ but not in controls. In the subsequent updated exome study of 4877 patients with SCZ and 6203 controls showed that there was one control subject with p. P170L (c.509 C>T) in CDK5 but no missense or nonsense variants in patients with SCZ. R50W was later found in the largest exome database, the Exome Aggregation Consortium. We further combined the data from the largest SCZ exome study and our study to examine whether the number of rare variants in CDK5 was significantly different between SCZ patients and controls, as both studies referred to rare variants with MAF<0.5%. The total number of rare variants within CDK5 was not statistically significant (P=0.179, Fisher’s exact test). Additional research using a larger sample size may be required, as the races and pipeline for detecting the rare variants were different between the studies.
Recently, excessive dopamine D2 receptor activation during early adolescence was shown to prevent the development of dendritic spines and led to cognitive dysfunction, a core symptom of SCZ, in an NR2B-dependent manner.55 Thus, the dysregulation of the composition of NMDARs may affect the dopamine levels during early adolescence, thereby affecting synapse maturation.56 Therefore, the involvement of other novel switching regulator genes warrants further research.
In conclusion, we examined whether the developmental NMDAR subunit switch was genetically involved in the pathophysiology of SCZ by re-sequencing three genes that regulate the composition of NMDARs. We found four novel variants in CDK5 in patients with SCZ but not in controls, although the difference did not show statistical significance. Additional research using a larger sample size or examining other switching regulator genes is warranted.
Additional infomation
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Lewis DA, Levitt P . Schizophrenia as a disorder of neurodevelopment. Ann Rev Neurosci 2002; 25: 409–432.
Lin Y-C, Koleske AJ . Mechanisms of synapse and dendrite maintenance and their disruption in psychiatric and neurodegenerative disorders. Ann Rev Neurosci 2010; 33: 349.
Luna B, Sweeney JA . Studies of brain and cognitive maturation through childhood and adolescence: a strategy for testing neurodevelopmental hypotheses. Schizophr Bull 2001; 27: 443–455.
Jentsch JD, Roth RH . The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology 1999; 20: 201–225.
Malhotra AK, Pinals DA, Weingartner H, Sirocco K, Missar CD, Pickar D et al. NMDA receptor function and human cognition: the effects of ketamine in healthy volunteers. Neuropsychopharmacology 1996; 14: 301–307.
Gao X-M, Sakai K, Roberts RC, Conley RR, Dean B, Tamminga CA . Ionotropic glutamate receptors and expression of N-methyl-D-aspartate receptor subunits in subregions of human hippocampus: effects of schizophrenia. Am J Psychiatry 2000; 157: 1141–1149.
Svensson TH . Dysfunctional brain dopamine systems induced by psychotomimetic NMDA-receptor antagonists and the effects of antipsychotic drugs. Brain Res Rev 2000; 31: 320–329.
Kapur S, Seeman P . NMDA receptor antagonists ketamine and PCP have direct effects on the dopamine D (2) and serotonin 5-HT (2) receptors-implications for models of schizophrenia. Mol Psychiatry 2001; 7: 837–844.
Iwayama-Shigeno Y, Yamada K, Itokawa M, Toyota T, Meerabux J, Minabe Y et al. Extended analyses support the association of a functional (GT) n polymorphism in the GRIN2A promoter with Japanese schizophrenia. Neurosci Lett 2005; 378: 102–105.
Itokawa M, Yamada K, Yoshitsugu K, Toyota T, Suga T, Ohba H et al. A microsatellite repeat in the promoter of the N-methyl-D-aspartate receptor 2A subunit (GRIN2A) gene suppresses transcriptional activity and correlates with chronic outcome in schizophrenia. Pharmacogenet Genomics 2003; 13: 271–278.
Tang J, Chen X, Xu X, Wu R, Zhao J, Hu Z et al. Significant linkage and association between a functional (GT) n polymorphism in promoter of the N-methyl-D-aspartate receptor subunit gene (GRIN2A) and schizophrenia. Neurosci Lett 2006; 409: 80–82.
Javitt DC, Zukin SR . Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry 1991; 148: 1301–1308.
Consortium SWGotPG. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014; 511: 421–427.
Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 2014; 506: 179–184.
Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 2014; 506: 185–190.
Cull-Candy S, Brickley S, Farrant M . NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol 2001; 11: 327–335.
Paoletti P, Bellone C, Zhou Q . NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 2013; 14: 383–400.
Sanz-Clemente A, Nicoll RA, Roche KW . Diversity in NMDA receptor composition many regulators, many consequences. Neuroscientist 2013; 19: 62–75.
Gambrill AC, Barria A . NMDA receptor subunit composition controls synaptogenesis and synapse stabilization. Proc Natl Acad Sci 2011; 108: 5855–5860.
Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW et al. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci 2007; 27: 2846–2857.
Yamakura T, Shimoji K . Subunit-and site-specific pharmacology of the NMDA receptor channel. Prog Neurobiol 1999; 59: 279–298.
Gielen M, Retchless BS, Mony L, Johnson JW, Paoletti P . Mechanism of differential control of NMDA receptor activity by NR2 subunits. Nature 2009; 459: 703–707.
Williams K, Russell SL, Shen YM, Molinoff PB . Developmental switch in the expression of NMDA receptors occurs in vivo and in vitro. Neuron 1993; 10: 267–278.
Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH . Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 1994; 12: 529–540.
Zhang Z, Sun QQ . Development of NMDA NR2 subunits and their roles in critical period maturation of neocortical GABAergic interneurons. Dev Neurobiol 2011; 71: 221–245.
Sanz-Clemente A, Matta JA, Isaac JT, Roche KW . Casein kinase 2 regulates the NR2 subunit composition of synaptic NMDA receptors. Neuron 2010; 67: 984–996.
Zhang S, Edelmann L, Liu J, Crandall JE, Morabito MA . Cdk5 regulates the phosphorylation of tyrosine 1472 NR2B and the surface expression of NMDA receptors. J Neurosci 2008; 28: 415–424.
Henderson JT, Georgiou J, Jia Z, Robertson J, Elowe S, Roder JC et al. The receptor tyrosine kinase EphB2 regulates NMDA-dependent synaptic function. Neuron 2001; 32: 1041–1056.
Nolt MJ, Lin Y, Hruska M, Murphy J, Sheffler-Colins SI, Kayser MS et al. EphB controls NMDA receptor function and synaptic targeting in a subunit-specific manner. J Neurosci 2011; 31: 5353–5364.
Tsai L-H, Takahashi T, Caviness V, Harlow E . Activity and expression pattern of cyclin-dependent kinase 5 in the embryonic mouse nervous system. Development 1993; 119: 1029–1040.
Chaperon F, Müller W, Auberson Y, Tricklebank M, Neijt H . Substitution for PCP, disruption of prepulse inhibition and hyperactivity induced by N-methyl-D-aspartate receptor antagonists: preferential involvement of the NR2B rather than NR2A subunit. Behav Pharmacol 2003; 14: 477–487.
Farber NB, Wozniak DF, Price MT, Labruyere J, Huss J, St Peter H et al. Age-specific neurotoxicity in the rat associated with NMDA receptor blockade: potential relevance to schizophrenia? Biol Psychiatry 1995; 38: 788–796.
Clinton SM, Haroutunian V, Meador-Woodruff JH . Up-regulation of NMDA receptor subunit and post-synaptic density protein expression in the thalamus of elderly patients with schizophrenia. J Neurochem 2006; 98: 1114–1125.
Grimwood S, Slater P, Deakin JFW, Hutson PH . NR2B-containing NMDA receptors are up-regulated in temporal cortex in schizophrenia. Neuroreport 1999; 10: 461–465.
Bitanihirwe B, Lim M, Kelley J, Kaneko T, Woo T . Glutamatergic deficits and parvalbumin-containing inhibitory neurons in the prefrontal cortex in schizophrenia. BMC Psychiatry 2009; 9: 71.
Ramos-Miguel A, Meana JJ, García-Sevilla JA . Cyclin-dependent kinase-5 and p35/p25 activators in schizophrenia and major depression prefrontal cortex: basal contents and effects of psychotropic medications. Int J Neuropsychopharmacol 2013; 16: 683–689.
Engmann O, Hortobágyi T, Pidsley R, Troakes C, Bernstein H-G, Kreutz MR et al. Schizophrenia is associated with dysregulation of a Cdk5 activator that regulates synaptic protein expression and cognition. Brain 2011; 134: 2408–2421.
Aksenova M, Burbaeva GS, Kandror K, Kapkov D, Stepanov A . The decreased level of casein kinase 2 in brain cortex of schizophrenic and Alzheimer's disease patients. FEBS Lett 1991; 279: 55–57.
Jeong J, Park Y-U, Kim D-K, Lee S, Kwak Y, Lee S-A et al. Cdk5 phosphorylates dopamine d2 receptor and attenuates downstream signaling. PLoS ONE 2013; 8: e84482.
Lai C-Y, Yu S-L, Hsieh MH, Chen C-H, Chen H-Y, Wen C-C et al. MicroRNA expression aberration as potential peripheral blood biomarkers for schizophrenia. PLoS ONE 2011; 6: e21635.
Segnitz N, Ferbert T, Schmitt A, Gass P, Gebicke-Haerter PJ, Zink M . Effects of chronic oral treatment with aripiprazole on the expression of NMDA receptor subunits and binding sites in rat brain. Psychopharmacology 2011; 217: 127–142.
Penzes P, Jones KA . Dendritic spine dynamics-a key role for kalirin-7. Trends Neurosci 2008; 31: 419–427.
Lee SH, DeCandia TR, Ripke S, Yang J, Sullivan PF, Goddard ME et al. Estimating the proportion of variation in susceptibility to schizophrenia captured by common SNPs. Nat Genet 2012; 44: 247–250.
Owen MJ, Craddock N, O’donovan MC . Suggestion of roles for both common and rare risk variants in genome-wide studies of schizophrenia. Arch Gen Psychiatry 2010; 67: 667–673.
Sheehan DV, Lecrubier Y, Sheehan KH, Amorim P, Janavs J, Weiller E et al. The Mini-International Neuropsychiatric Interview (MINI): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry 1998; 59: 22–33.
Miller S, Dykes D, Polesky H . A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988; 16: 1215.
Lepri FR, Scavelli R, Digilio MC, Gnazzo M, Grotta S, Dentici ML et al. Diagnosis of Noonan syndrome and related disorders using target next generation sequencing. BMC Med Genet 2014; 15: 14.
Li H, Durbin R . Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–1760.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al. The sequence alignment/map format and SAMtools. Bioinformatics 2009; 25: 2078–2079.
Liu X, Jian X, Boerwinkle E . dbNSFP v2. 0: A database of human non-synonymous SNVs and their functional predictions and annotations. Hum Mut 2013; 34: E2393–E2402.
Mapelli M, Musacchio A . The structural perspective on CDK5. Neurosignals 2003; 12: 164–172.
Tarricone C, Dhavan R, Peng J, Areces LB, Tsai L-H, Musacchio A . Structure and regulation of the CDK5–p25 nck5a complex. Mol Cell 2001; 8: 657–669.
Dhavan R, Tsai L-H . A decade of CDK5. Nat Rev Mol Cell Biol 2001; 2: 749–759.
Maccioni RB, Otth C, Concha II, Munoz JP . The protein kinase Cdk5. FEBS J 2001; 268: 1518–1527.
Jia J-M, Zhao J, Hu Z, Lindberg D, Li Z . Age-dependent regulation of synaptic connections by dopamine D2 receptors. Nat Neurosci 2013; 16: 1627–1636.
Genovese G, Fromer M, Stahl EA, Ruderfer DM, Chambert K, Landén M et al. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nat Neurosci 2016; 19: 1433–1441.
Acknowledgements
The present study was supported by a grant awarded to KK from the Development of Biomarker Candidates for Social Behavior Study, which was carried out under the Strategic Research Program for Brain Sciences by the Ministry of Education, Culture, Sports, Science, and Technology of Japan. This study was supported in part by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Numbers JP16H06395, 16H06399 and 16K21720 to KK and in part by the Brain Mapping by Integrated Neurotechnologies for Disease Studies (Brain/MINDS) from the Japan Agency for Medical Research and Development, AMED, to KK. We are grateful for the generosity of the patients and their families, healthy control participants, and all of the medical staff and researchers who cooperated in this work.
Author information
Authors and Affiliations
Contributions
AY and CK designed the study. AY, FN, and KK performed the experiments. AY undertook the statistical analysis and wrote the manuscript. All of the authors obtained blood samples from patients and healthy subjects. All authors have approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information for this article can be found on the Human Genome Variation website .
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Yoshikawa, A., Nishimura, F., Inai, A. et al. Novel rare variations in genes that regulate developmental change in N-methyl-d-aspartate receptor in patients with schizophrenia. Hum Genome Var 5, 17056 (2018). https://doi.org/10.1038/hgv.2017.56
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2017.56
This article is cited by
-
Sex-dependent role for EPHB2 in brain development and autism-associated behavior
Neuropsychopharmacology (2021)
