
Abstract
Malan syndrome has recently been characterized to present Sotos-like phenotypes, such as intellectual disability and macrocephaly, with mutations in the NFIX gene. Herein, we report a 38-year-old patient with a novel single adenine insertion mutation in exon 2 of the NFIX gene (c.290_291insA). He developed early-onset thoracic aortic aneurysm and dissection, which was a rare complication but deserves particular attention in relatively long-lived patients with Sotos-like phenotypes.
Similar content being viewed by others
Sotos syndrome is a rare, autosomal dominant, multisystemic genetic disorder characterized by distinctive facial features, intellectual disability, and overgrowth of the body in early life with macrocephaly. In ~90% of Sotos syndrome cases, mutations occur in the gene encoding nuclear receptor-binding SET domain containing protein 1 (NSD1),1, 2,3 located on chromosome 5q35. Recently, the NFIX gene on chromosome 19p13, which encodes nuclear factor I/X, was reported as a causative gene for Sotos-like phenotypes (known as Sotos syndrome 2 or Malan syndrome (OMIM #614753)),4,5, 6 and a total of such 33 patients aged 1–27 years have been reported. Most cases have been sporadic owing to the severity of the phenotype. Since mutations in the NFIX gene are known to cause Marshall–Smith syndrome (MSS (OMIM #602535)), which is characterized by the triad of facial dysmorphism, failure to thrive, and accelerated osseous maturation, the clinical and genetic differences between these two NFIX-associated diseases have been actively explored.
Herein, we identified a novel single-base adenine (A) insertion mutation in the coding region NFIX gene in a Japanese patient with Sotos-like phenotypes. He had early-onset thoracic aortic aneurysm with mild aortic regurgitation and developed thoracic aortic dissection at the age of 38 years. Although a mildly dilated aortic root at the sinus of Valsalva was previously reported in an 11-year-old Caucasian American girl who was carrying a 19p13.13 deletion disrupting NFIX and CACNA1A,7 there have been no published cases complicated with aortic dissection in patients with Sotos-like phenotypes. Our patient is the oldest reported patient with NFIX mutations, and thoracic aortic aneurysm and dissection (TAAD) deserve particular attention in relatively long-lived patients with Sotos-like phenotypes.
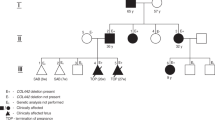
The proband (No. 882) is the first son of healthy cousin parents (No. 883, No. 884) (Figure 1a). During the pregnancy, macrocephaly was suspected, and he was born at week 41 of gestation by cesarean section due to breech position. The Apgar score was 10 out of 10 at one minute after birth, and his body weight was 3400 g (50–75th percentile), height was 50 cm (50–75th percentile), and head circumference (HC) was 38.5 cm (>97th percentile) (Figure 1b,c). During his development, delayed speech and unsmooth gait gradually became obvious, and his HC remained greater than 2 s.d. above the mean (Figure 1c). His initial attack of recurrent convulsive seizures happened when he was 2 years old, and he was admitted at the age of 4.5 years for our first examination. His weight was 18.7 kg (+1SD), height was 114.5 cm (+2.4 SD), and HC was 54.2 cm (+2.3 SD). Carpal radiograph showed a bone age of 6 years. Abnormal phenotype included intellectual disability, recurrent convulsive seizures, excessive growth of the body, large hands and feet, cubitus valgus, crus varum, advanced bone age, funnel chest, kyphoscoliosis, tendon hyperreflexia (biceps, patellar, Achilles), and distinctive facial features, including macrocephaly, prominent forehead, hypertelorism, down-slanting eyes, prognathism, and high narrow palate; thus, Sotos syndrome was suspected. Until recently, he had been followed up by pediatricians and psychiatrists under treatment with anticonvulsants without unplanned admissions.

Japanese case of Malan syndrome with aortic aneurysm and dissection. (a) Pedigree of a Japanese case of Malan syndrome. The age is shown in the upper left corner. The plus and minus symbols in the upper right corner indicate individuals with and without the mutation, respectively. Square, male; circle, female; arrow, proband; diagonal line, died. (b, c) Height (B, red circles), weight (b, blue circles) and head circumference (c, red circles) chart with the reference lines showing the 3th, 10th, 25th, 50th, 75th, 90th, and 97th percentiles for Japanese boy. (d, e) Transthoracic echocardiography in parasternal long-axis view showing a mildly dilated ascending aorta (39 mm) at the age of 35 years (d), and a progressive dilatation of the ascending aorta (66 mm) with proximal dissecting flap (arrowhead) at the age of 38 years (e), respectively. LA, left atrium; LV, left ventricle; Ao, aorta. (f) Enhanced computed tomography showing dilated aortic root with dissection.
He was referred to a hypertension specialist at the age of 32 years for blood pressure increase to over 200 mm Hg during seizures. However, the blood pressure at home was within normal limits, and secondary causes of hypertension were excluded. At the age of 35 years, electrocardiogram on annual medical check-up showed increased voltage (SV1+RV5=4.1 mV). Transthoracic echocardiography (TTE) revealed the absence of left ventricular hypertrophy but the presence of a mildly dilated ascending aorta (39 mm) (Figure 1d), and the annual check-ups continued. At the age of 38 years, his weight was 74 kg, height was 175 cm, HC was 61.4 cm (+4.4 SD), and he did not have diabetes, dyslipidemia, or complaints of aches and pains during the most recent one-year period. However, TTE showed a progressive dilatation of the ascending aorta (66 mm) with proximal dissecting flap (Figure 1e), which extended into the brachiocephalic artery, as evaluated by computed tomography (Figure 1f). Since he had passed the acute phase and it was determined that surgical repair would be difficult for psychiatric and social reasons, he has been carefully followed up by a cardiologist under treatment with antihypertensive drugs.
For the proband and his parents, we performed genetic analysis of TAAD and Sotos syndrome-related gene mutations, as previously described.8 This investigation was approved by the University of Tokyo Hospital ethics committee (G-1538). The fluorescence in situ hybridization (FISH) test did not detect a deletion or duplication of 5q35 encompassing the NSD1 gene (SRL Co, Tokyo, Japan), and we did not perform an array CGH (comparative genomic hybridization) test for chromosome abnormalities. Sanger DNA sequence of the NSD1, NFIX, FBN1, TGFBR1, TGFBR2, SMAD3, TGFB2, TGFB3, MYH11, and ACTA2 genes revealed a novel single adenine insertion mutation (c.290_291insA, p.Asp98GlyfsX29) within exon 2 of the NFIX gene (NM_001271043; NP_001257972) in the proband (Figure 2), but not in his parents (data not shown). This variant was confirmed by the direct-sequencing of subcloned PCR products spanning this variant (Figure 2b), and is currently not listed in public databases of variants, such as dbSNP (http://www.ncbi.nlm.nih.gov/SNP/), Exome Variant Server (EVS) (http://evs.gs.washington.edu/EVS/), Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org), or Human Genetic Variation Browser (http://www.genome.med.kyoto-u.ac.jp/SnpDB/), and there were no mutations in the NSD1 gene or well-known TAAD-related genes. Thus, we concluded that this de novo mutation of the NFIX gene was possibly responsible for the Sotos-like phenotypes. The patient was finally diagnosed with Malan syndrome based on his comprehensive clinical features and the result of genetic analysis.

NFIX mutation analysis. (a) Genome DNA sequencing identified a frameshift mutation in exon 2 of the NFIX gene in the proband #882 (c.290_291insA). (b) Sequence analyses of the subcloned PCR products amplified from genomic DNA of the proband. PCR was performed using primers spanning the variant: forward, 5′-CCCCTTCTAACGCTGCTTTT -3′ and reverse, 5′-ATCACCATGACCAGGTCCAG-3′. The products were then subcloned into a pMD20T vector using a Mighty TA-cloning Kit (TaKaRa, Shiga, Japan) and sequenced. Predicted amino acid sequences and numbers are shown along with nucleotide sequences. Aspartate (Asp) at amino acid positon 98 is the first amino acid changed in the mutated allele, predicting a premature stop mutation at codon 126 (p.Asp98GlyfsX29).
Mutations in the NFIX gene can cause two distinct diseases; most mutations causing Malan syndrome cluster in the evolutionally conserved N-terminal DNA-binding/dimerization domain (exons 2 and 3), and mutations associated with Marshall–Smith syndrome cluster in the C-terminal end (exons 6 to 10).4,5, 6 Malan et al.4 speculated that differences in phenotype consequences are due to differences in the fate of the mRNAs; the haploinsufficiency due to nonsense-mediated mRNA decay (NMD) and the dominant-negative effect of putative truncated proteins likely lead to Malan syndrome and Marshall–Smith syndrome, respectively. Consistent with these observations, in the present case, the single-base adenine (A) insertion mutation within exon 2 produced frameshift and a premature termination codon, and thus, the mutant mRNAs are presumably degradated by NMD.
The roles of transcription factor NFIX are becoming evident, particularly in the development of the brain, skeletal muscle, and hematopoietic system;9,10,11,12 however, the pathophysiological roles of NFIX in vessels remain elusive. Nimmakayalu et al. previously reported an 11-year-old Caucasian American girl with a 19p13.13 deletion that disrupted NFIX and CACNA1A due to parental germline mosaicism and led to macrocephaly, intellectual disability, episodic ataxia, and a mildly dilated aortic root at the sinus of Valsalva (29 mm; Z-score=3.4).7 In contrast, the elder sister aged 16 years with the same chromosome deletion had macrocephaly and intellectual disability but no aortic dilatation. Among 33 previously reported young patients with Malan syndrome (age 1–27 years), TAADs have not been yet reported, and thus, our case is the oldest case and the second case of aortopathy among patients with NFIX mutation-related Sotos-like syndrome. It is possible that the phenotype is influenced by other genetic and/or environmental modifiers of aortopathy, and further investigations are needed to better define the aortic phenotype in patients with NFIX mutations.
In conclusion, we report a sporadic Japanese case of Malan syndrome with a novel NFIX mutation (c.290_291insA, p.Asp98GlyfsX29), which caused Sotos-like phenotypes complicated with early-onset TAAD; however, the exact mechanism underlying this association remains unclear. Evaluation of the thoracic aorta and genetic testing in patients with Sotos-like phenotypes may contribute to the detection of such high-risk subgroups that require improved risk-stratification and management.
References
References
Douglas J, Hanks S, Temple IK, Davies S, Murray A, Upadhyaya M et al. NSD1 mutations are the major cause of Sotos syndrome and occur in some cases of Weaver syndrome but are rare in other overgrowth phenotypes. Am J Hum Genet 2003; 72: 132–143.
Turkmen S, Gillessen-Kaesbach G, Meinecke P, Albrecht B, Neumann LM, Hesse V et al. Mutations in NSD1 are responsible for Sotos syndrome, but are not a frequent finding in other overgrowth phenotypes. Eur J Hum Genet 2003; 11: 858–865.
Waggoner DJ, Raca G, Welch K, Dempsey M, Anderes E, Ostrovnaya I et al. NSD1 analysis for Sotos syndrome: insights and perspectives from the clinical laboratory. Genet Med 2005; 7: 524–533.
Malan V, Rajan D, Thomas S, Shaw AC, Louis Dit Picard H, Layet V et al. Distinct effects of allelic NFIX mutations on nonsense-mediated mRNA decay engender either a Sotos-like or a Marshall-Smith syndrome. Am J Hum Genet 2010; 87: 189–198.
Klaassens M, Morrogh D, Rosser EM, Jaffer F, Vreeburg M, Bok LA et al. Malan syndrome: Sotos-like overgrowth with de novo NFIX sequence variants and deletions in six new patients and a review of the literature. Eur J Hum Genet 2015; 23: 610–615.
Martinez F, Marin-Reina P, Sanchis-Calvo A, Perez-Aytes A, Oltra S, Rosello M et al. Novel mutations of NFIX gene causing Marshall-Smith syndrome or Sotos-like syndrome: one gene, two phenotypes. Pediatr Res 2015; 78: 533–539.
Nimmakayalu M, Horton VK, Darbro B, Patil SR, Alsayouf H, Keppler-Noreuil K et al. Apparent germline mosaicism for a novel 19p13.13 deletion disrupting NFIX and CACNA1A. Am J Med Genet A 2013; 161A: 1105–1109.
Takeda N, Morita H, Fujita D, Inuzuka R, Taniguchi Y, Nawata K et al. A deleterious MYH11 mutation causing familial thoracic aortic dissection. Hum Genome Var 2015; 2: 15028.
Driller K, Pagenstecher A, Uhl M, Omran H, Berlis A, Grunder A et al. Nuclear factor I X deficiency causes brain malformation and severe skeletal defects. Mol Cell Biol 2007; 27: 3855–3867.
Campbell CE, Piper M, Plachez C, Yeh YT, Baizer JS, Osinski JM et al. The transcription factor Nfix is essential for normal brain development. BMC Dev Biol 2008; 8: 52.
Rossi G, Antonini S, Bonfanti C, Monteverde S, Vezzali C, Tajbakhsh S et al. Nfix regulates temporal progression of muscle regeneration through modulation of myostatin expression. Cell Rep 2016; 14: 2238–2249.
Holmfeldt P, Pardieck J, Saulsberry AC, Nandakumar SK, Finkelstein D, Gray JT et al. Nfix is a novel regulator of murine hematopoietic stem and progenitor cell survival. Blood 2013; 122: 2987–2996.
Data Citations
Takeda, Norifumi HGV Database (2017) http://dx.doi.org/10.6084/m9.figshare.hgv.1367
Acknowledgements
We thank Ms Yuko Suzuki for her excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Oshima, T., Hara, H., Takeda, N. et al. A novel mutation of NFIX causes Sotos-like syndrome (Malan syndrome) complicated with thoracic aortic aneurysm and dissection. Hum Genome Var 4, 17022 (2017). https://doi.org/10.1038/hgv.2017.22
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2017.22
This article is cited by
-
A deep phenotyping experience: up to date in management and diagnosis of Malan syndrome in a single center surveillance report
Orphanet Journal of Rare Diseases (2022)
