
Abstract
Smith–Lemli–Opitz syndrome is an autosomal recessive disease caused by mutations in 7-dehydrocholesterol reductase (DHCR7), which is rarely observed in Japan. We report a Japanese case with 46,XY disorder of sex development and Y-shaped 2–3 toe syndactyly. DHCR7 gene analysis revealed compound heterozygous mutations including the novel mutation H442R. Early diagnosis led to starting cholesterol treatment at an early age.
Similar content being viewed by others

Smith–Lemli–Opitz syndrome (SLOS, OMIM #270400) is an autosomal recessive inheritance disease characterized by multiple anomaly, failure to thrive and intellectual disability.1–3 SLOS is caused by defective function of 7-dehydrocholesterol reductase (DHCR7), which converts 7-dehydrocholesterol (7-DHC) to cholesterol.4 Thus far, over 140 DHCR7 mutations have been reported.5,6
Because of defective cholesterol synthesis, serum cholesterol levels are reduced in patients, and 7-DHC and 8-dehydrocholesterol tend to accumulate. These abnormalities in cholesterol synthesis may cause various problems during the fetal period, including dysmorphic facial features, hypotonicity, microcephaly, Y-shaped 2–3 toe syndactyly, 46,XY disorder of sex development (DSD) and congenital heart diseases.3 Y-shaped 2–3 toe syndactyly, referring to incomplete cutaneous syndactyly of the second and third toes, is a unique SLOS symptom.2 However, the other phenotypes show a wide spectrum of severity, ranging from mild to severe in nature. Clinical severity is thought to depend on the extent that the mutation impairs DHCR7 function.5
Cholesterol treatment is beneficial for neurological development and neuropsychological symptom treatment, and is widely used in Europe.7,8 However, the reports of cholesterol treatment for SLOS in Japan are limited. We report a Japanese SLOS case with a novel mutation in DHCR7 who was diagnosed early in life with 46,XY DSD and Y-shaped 2–3 toe syndactyly, and was treated with cholesterol.
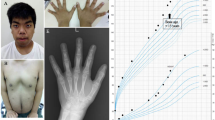
The patient was born at 38 weeks of gestation to non-consanguineous Japanese parents. Birth weight, length and head circumference were 2,595 g (−0.92 s.d.), 45 cm (−1.7 s.d.) and 33 cm (−0.1 s.d.), respectively. Ambiguous genitalia, low set ears, a short nose and Y-shaped 2–3 toe syndactyly were detected at birth (Figure 1a and b). Examination of the patient’s genitalia revealed a micropenis (stretch length: 1.5 cm), left lateral testicular hypoplasia (right and left diameters: 10 and 5 mm, respectively), hypospadias and a bifid scrotum (Figure 1c). Differential diagnosis of DSD was performed. Echography and magnetic resonance imaging failed to detect the presence of a uterus or ovaries. Blood examination revealed a relatively low total cholesterol (T. Chol) level (46 mg/dl) (reference: 129–232), normal testosterone and slightly low anti-mullerian hormone level for males. Urine steroid profile for detection of defects in steroid hormone and testosterone synthesis was normal. G-banding analysis confirmed a normal 46,XY male karyotype. From these results, the patient was recorded in public office documents as male. The 46,XY DSD and the observation that his phenotype included syndactyly suggested that he had SLOS. We performed cholesterol analysis with high-performance liquid chromatography. This analysis revealed relative hypocholesterolemia and high 7-DHC and 8-dehydrocholesterol levels in the patient and normal levels (undetectable) in his parents (Table 1). T. Chol/7-DHC was significantly lower in the patient than in his parents. Therefore, he was biochemically diagnosed with SLOS. The patient also presented with allergies for milk and eggs, and eczema throughout his body. His eczema, considered to be atopic dermatitis, was treated with corticosteroid ointment.

Clinical appearance of the patient. (a, b) Bilateral Y-shaped 2–3 toe syndactyly. (c) Ambiguous genitalia, including micropenis, left lateral testicular hypoplasia, hypospadias and a bifid scrotum. (d) Chart depicting the cholesterol dose and serum T. Chol level. (e) Growth curve showing development after treatment initiation. T. Chol, total cholesterol.
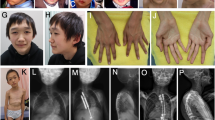
To reveal the genetic background of his disease, we performed DHCR7 gene analysis of the patient (age: 2 months) and his parents. We obtained written informed consent from the parents for DNA analysis and approval from the ethics committee of the University of Tokyo. The analysis revealed two heterozygous missense mutations, c.907G>A, p.Gly303Arg (G303R) and c.1325A>G, p.His442Arg (H442R) (Figure 2a). G303R, located on exon 8, had been reported in a Japanese SLOS case.9 However, H442R represented a novel missense mutation located on exon 9 and was not found in an single-nucleotide polymorphism database of 934 Japanese people nor in the single-nucleotide polymorphism database of the 1,000 Genome Project.10 In silico functional prediction using PolyPhen 211 and Mutation Taster12 indicated that the H442R substitution was ‘probably damaging’ and ‘disease causing’, respectively. Genetic analysis of the parents revealed that his father and mother carried G303R and H442R, respectively, in a heterozygous manner (Figure 2a).

DHCR7 gene analysis. (a) The patient exhibited two non-synonymous substitutions. G303R, inherited from his father, has been reported previously in Japanese cases; H442R, inherited from his mother, has not been reported previously nor deposited in an established SNP database. Genetic tests showed that both parents were carriers. (b) Homologs of DHCR7 at the H442 residue, which is highly conserved among different species. (c) DHCR7 gene structure. Boxes indicate exons and lines indicate introns. H442R was located in a region where other missense and nonsense mutations exist. SNP, single-nucleotide polymorphism.
Although oral cholesterol is widely used in Europe, cholesterol powder is not an approved medical drug in Japan. However, for our patient, we obtained approval from the ethics committee of Tokyo University and informed consent from his parents to begin cholesterol treatment at 3 months of age. There are several forms of cholesterol treatment, including cholesterol suspension, yolk oils and egg yolk. Here, we selected cholesterol suspension because of the patient’s food allergies. After 3 months of treatment, the T. Chol level increased to the normal range, and the 7-DHC level reduced from 8.0 to 3.8 mg/dl. The cholesterol dose was gradually increased from 50 to 85 mg/kg/day at 2 years (Figure 1d). His height gain followed approximately the −3 s.d. line of the normal growth curve (Figure 1e). Although many SLOS cases exhibit microcephaly, his head circumference also remained at −3 s.d., which is not small compared to his height. At 2 years, his motor and language development, and his social development had reached that normally expected at 1 and 1.5 years of age, respectively. Eczema is often seen in SLOS patients. This patient also had refractory eczema, which was ameliorated after cholesterol treatment together with topical corticosteroid ointment.
We identified a novel mutation H442R of DHCR7 in a patient with a mild SLOS phenotype. This substitution was not recorded in the single-nucleotide polymorphism database. The H442 residue is highly conserved in mouse, chicken and zebrafish (Figure 2b); in silico analysis shows that mutations of this residue are predicted to damage protein function. DHCR7 is composed of 9 exons, and the predicted structure of the protein contains 9 membrane-associated helixes (MAH).13 H442 is located in exon 9 at the C-terminus (between membrane-associated helixes 9 and the 3′untranslated region), the area where other missense mutations have been reported (Figure 2c). The phenotype of a patient with one mutation in the C-terminus region is reportedly mild.13 Considering the mild phenotype of our patient, we suspected that DHCR7 with the H442R mutation exhibited residual enzyme activity. The other mutation, G303R, has been reported in several Japanese cases and is located in exon 8, encoding loop 6–7 within the sterol sensing domain.14 Symptoms of cases possessing G303R as a compound heterozygote usually present with mild-to-severe phenotypes.9 Therefore, we believe that the compound heterozygous mutation caused SLOS.
SLOS incidence is estimated as 1:20,000–1:60,000 in Europe, but it is lower in other areas and is extremely rare in Japan.2 Genital anomalies are seen in more than 70% of SLOS patients.2 Although the mechanism of genital anomalies in SLOS has not been elucidated, it is suggested that it might be caused by the lack of substrate to produce adrenal and testicular steroids owing to low-cholesterol synthesis.15 Because the phenotype occurs before birth, cholesterol treatment after birth is unlikely to be beneficial for the genital anomalies. However, early diagnosis and treatment of SLOS is important because cholesterol treatment appears to improve physical and neurological development.7,8 In our case, we diagnosed SLOS by differential diagnosis of 46,XY DSD and subsequently confirmed our suspicions by cholesterol analysis and DHCR7 gene analysis (at 2 months). This enabled treatment at the early age of 3 months. Even though SLOS is a very rare syndrome, it should be considered in a differential diagnosis of 46,XY DSD.
In SLOS, cholesterol levels vary. Calculating T. Chol/7-DHC allows differentiation between affected (0.1–10) and healthy individuals (>10,000).16 In our case, the T. Chol level was relatively low, and T. Chol/7-DHC was definitively low compared with that of healthy controls. Consequently, this important ratio was very useful for diagnosing SLOS. The 7-DHC level, which is reported to correlate with severity, was 8.0 mg/dl before treatment. Other reported Japanese cases showed higher 7-DHC levels (9.7–35 mg/dl).9 Considering clinical, genetic and laboratory data, this case appeared to be mild compared with other reported cases. Although cholesterol analysis by high-performance liquid chromatography led to the definitive diagnosis of SLOS, it can be conducted only in research institutes. We consider genetic analysis to be more applicable for diagnosis.
Dietary cholesterol treatment is broadly used as a treatment for SLOS worldwide and results in an increase in the serum cholesterol level and decrease in 7-DHC and 8-dehydrocholesterol levels by feedback inhibition.7 Elias et al.17 reported that neuropsychological symptoms were ameliorated rapidly with cholesterol treatment. Although our patient had shown postnatal growth failure before treatment, he has maintained growth after cholesterol treatment and has not shown a decline in development in either microcephaly or other neurological symptoms. However, the extent to which cholesterol treatment has contributed to his growth and neurological development is unclear because the natural course of SLOS varies and he was classified to have only mild severity.
In summary, we report a Japanese SLOS case with a novel DHCR7 mutation, who presented with 46,XY DSD, Y-shaped 2–3 toe syndactyly and a low-T. Chol/7-DHC ratio, and who was treated with cholesterol. SLOS should be considered as a potential differential diagnosis of 46,XY DSD because early diagnosis leads to early treatment.
Accession codes
Nucleotide sequence data reported are available in the DDBJ/EMBL/GenBank databases under the accession numbers: LC201749, LC202815 and LC202816.
References
References
Smith DW, Lemli L, Opitz JM . A newly recognized syndrome of multiple congenital anomalies. J Pediatr 1964; 64: 210–217.
Kelley RI, Hennekam RC . The Smith-Lemli-Opitz syndrome. J Med Genet 2000; 37: 321–335.
Nowaczyk MJ, Irons MB . Smith-Lemli-Opitz syndrome: phenotype, natural history, and epidemiology. Am J Med Genet C Semin Med Genet 2012; 160C: 250–262.
Fitzky BU, Witsch-Baumgartner M, Erdel M, Lee JN, Paik YK, Glossmann H et al. Mutations in the Delta7-sterol reductase gene in patients with the Smith-Lemli-Opitz syndrome. Proc Natl Acad Sci USA 1998; 95: 8181–8186.
Witsch-Baumgartner M, Fitzky BU, Ogorelkova M, Kraft HG, Moebius FF, Glossmann H et al. Mutational spectrum in the Delta7-sterol reductase gene and genotype-phenotype correlation in 84 patients with Smith-Lemli-Opitz syndrome. Am J Hum Genet 2000; 66: 402–412.
Wassif CA, Krakowiak PA, Wright BS, Gewandter JS, Sterner AL, Javitt N et al. Residual cholesterol synthesis and simvastatin induction of cholesterol synthesis in Smith-Lemli-Opitz syndrome fibroblasts. Mol Genet Metab 2005; 85: 96–107.
Svoboda MD, Christie JM, Eroglu Y, Freeman KA, Steiner RD . Treatment of Smith-Lemli-Opitz syndrome and other sterol disorders. Am J Med Genet C Semin Med Genet 2012; 160C: 285–294.
Irons M, Elias ER, Abuelo D, Bull MJ, Greene CL, Johnson VP et al. Treatment of Smith-Lemli-Opitz syndrome: results of a multicenter trial. Am J Med Genet 1997; 68: 311–314.
Matsumoto Y, Morishima K, Honda A, Watabe S, Yamamoto M, Hara M et al. R352Q mutation of the DHCR7 gene is common among Japanese Smith-Lemli-Opitz syndrome patients. J Hum Genet 2005; 50: 353–356.
The Genomes Project C. A global reference for human genetic variation. Nature 2015; 526: 68–74.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P et al. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249.
Schwarz JM, Rodelsperger C, Schuelke M, Seelow D . MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010; 7: 575–576.
Waterham HR, Hennekam RC . Mutational spectrum of Smith-Lemli-Opitz syndrome. Am J Med Genet C Semin Med Genet 2012; 160C: 263–284.
Bae SH, Lee JN, Fitzky BU, Seong J, Paik YK . Cholesterol biosynthesis from lanosterol. Molecular cloning, tissue distribution, expression, chromosomal localization, and regulation of rat 7-dehydrocholesterol reductase, a Smith-Lemli-Opitz syndrome-related protein. J Biol Chem 1999; 274: 14624–14631.
Mendonca BB, Arnhold IJP, Domenice S, Costa EMF. 46,XY disorders of sexual development. In: De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM et al. (eds). Endotext. MDText.com: South Dartmouth, MA, USA, 2000.
Olah AV, Szabo GP, Varga J, Balogh L, Csabi G, Csakvary V et al. Relation between biomarkers and clinical severity in patients with Smith-Lemli-Opitz syndrome. Eur J Pediatr 2013; 172: 623–630.
Elias ER, Irons MB, Hurley AD, Tint GS, Salen G . Clinical effects of cholesterol supplementation in six patients with the Smith-Lemli-Opitz syndrome (SLOS). Am J Med Genet 1997; 68: 305–310.
Data Citations
Kitanaka, Sachiko HGV Database (2017) http://dx.doi.org/10.6084/m9.figshare.hgv.1348
Acknowledgements
We thank the patient and his parents for all their help and participation, and thank Reiko Onai for her technical support. This study was supported by a Grant-in-Aid from the Ministry of Education, Culture, Sports, Science, and Technology Japan (SK: 23591489), JSPS KAKENHI (to MT: JP16J04675).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Tamura, M., Isojima, T., Kasama, T. et al. Novel DHCR7 mutation in a case of Smith–Lemli–Opitz syndrome showing 46,XY disorder of sex development. Hum Genome Var 4, 17015 (2017). https://doi.org/10.1038/hgv.2017.15
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2017.15
