
Abstract
OCRL1 and its paralog INPP5B encode phosphatidylinositol 5-phosphatases that localize to the primary cilium and have roles in ciliogenesis. Mutations in OCRL1 cause the X-linked Dent disease type 2 (DD2; OMIM# 300555), characterized by low-molecular weight proteinuria, hypercalciuria, and the variable presence of cataracts, glaucoma and intellectual disability without structural brain anomalies. Disease-causing mutations in INPP5B have not been described in humans. Here, we report the case of an 11-year-old boy with short stature and an above-average IQ; severe proteinuria, hypercalciuria and osteopenia resulting in a vertebral compression fracture; and Chiari I malformation with cervico-thoracic syringohydromyelia requiring suboccipital decompression. Sequencing revealed a novel, de novo DD2-causing 462 bp deletion disrupting exon 3 of OCRL1 and a maternally inherited, extremely rare (ExAC allele frequency 8.4×10−6) damaging missense mutation in INPP5B (p.A51V). This mutation substitutes an evolutionarily conserved amino acid in the protein’s critical PH domain. In silico analyses of mutation impact predicted by SIFT, PolyPhen2, MetaSVM and CADD algorithms were all highly deleterious. Together, our findings report a novel association of DD2 with Chiari I malformation and syringohydromyelia, and document the effects of digenic mutation of human OCRL paralogs. These findings lend genetic support to the hypothesis that impaired ciliogenesis may contribute to the development of Chiari I malformation, and implicates OCRL-dependent PIP3 metabolism in this mechanism.
Similar content being viewed by others


Introduction
Dent disease is a rare X-linked recessive condition characterized by proximal tubule renal dysfunction leading to hypercalciuria, decreased renal tubular phosphate reabsorption, low-molecular weight proteinuria, aminoaciduria and variable presence of nephrolithiasis, nephrocalcinosis, hematuria and renal failure.1 Extrarenal manifestations may also be present, and include intellectual impairment, short stature, growth retardation and rickets in ~30% of patients.1–3 Many features of this condition are variable in severity, which led to initial reports of several separate disorders, sharing common characteristics which are collectively referred to as ’X-linked hypercalciuric nephrolithiasis.4–6
Advancements in genetic molecular screening methods have provided further insight into the genomic determinants that characterize this disease spectrum. Mutations in CLCN5 (Xp11.23) encoding a predominantly endosomal-bound Cl−/H+ exchanger, critically related to endosomal acidification, transepithelial transport and proximal tubule endocytosis, define Dent disease type 1.7 To date, ~200 mutations in CLCN5 have been reported in Dent disease patients,7 and account for ~50% of all cases.
Sequencing initiatives in CLCN5-negative patients with clinical features of Dent disease led to the discovery of mutations in OCRL1 (Xq26.1) in this population.3 While sharing the renal manifestations present in Dent disease type 1, patients harboring OCRL1 mutations display a different pattern of extrarenal manifestations, including variable cognitive impairment, subclinical cataracts and umbilical hernias. These phenotypic presentations coupled with OCRL1 mutations define Dent disease type 2 (DD2; OMIM #300555).8
OCRL1 was named due to the initial association with the Lowe Oculocerebrorenal Syndrome (OCRL; Lowe Syndrome; OMIM # 309000)7 which shares several of the phenotypic manifestations of DD2, but is more severe. Notably, there are several clinical differences between Lowe syndrome and DD2. Infants with Lowe syndrome are often born with bilateral congenital cataracts and other eye abnormalities that can impair vision. Approximately 50% of affected patients also develop infantile glaucoma with buphthalmos. Patients exhibit failure to thrive, neonatal hypotonia with resulting motor impairment, seizures and intellectual ability ranging from normal to severe mental retardation.9 Renal abnormalities, commonly Fanconi syndrome and hypophosphatemic rickets, are all characteristic of Lowe Syndrome.9
OCRL1 encodes a 5-phosphatase of phosphatidylinositol 4,5-bisphosphate, 1,4,5-trisphosphate and 1,3,4,5-tetrakisphosphate with subcellular localization at the plasma membrane, trans-Golgi complex and primary cilia.10–12 This protein has been linked to multiple key intracellular functions including vesicle trafficking, cytoskeleton stability due to α-actin distribution and ciliogenesis, in addition to intrinsic Rho GTPase binding.12
OCRL1 has one known human paralog, INPP5B, located on chromosome 1. OCRL1 and INPP5B share homology in the 5-phosphatase, abnormal spindle-like microcephaly associated protein (ASH) and rho GTPase-activating (RhoGAP) domains. The homology diverges in the pleckstrin homology (PH) domain where clathrin-binding sites in OCRL1 are absent in INPP5B.13 Knockout of Ocrl1 in mice (murine ortholog of OCRL1) failed to produce a phenotype. However, knockout of both Ocrl1 and Inpp5b resulted in embryonic lethality, suggesting compensatory functions of Ocrl1 by Inpp5b.14 Interestingly, an Ocrl1 and Inpp5b null mouse model expressing humanized INPP5B rescued the embryonic lethality phenotype and recapitulated the human DD2 phenotype characterized by low-molecular weight proteinuria and aminoaciduria with failure to thrive.15 Homozygous knockout of Inpp5B resulted in male infertility due to impaired spermatogenesis and decreased sperm mobility, which has been attributed to impaired cilia function.16 These studies suggest that while there may be some functional overlap between OCRL1 and INPP5B, the latter may also have distinct functions disparate from those of OCRL1 that have yet to be fully elucidated. To date, no mutations in INPP5B have been directly implicated in human disease.
Here, using whole-exome sequencing, we report the first patient with digenic mutations in paralogs OCRL1 and INPP5B resulting in a novel presentation of Dent’s disease type 2 with Chiari I malformation and syringohydromyelia.
Materials and methods
All procedures in this study comply with Yale University’s Human Investigation Committee and are Human Research Protection Program. Informed consent was obtained from all participants before sample collection.
Whole-exome sequencing and variant calling
Samples were sequenced at the Yale Center for Genome Analysis following the center's standard protocol. Targeted capture was performed using the Nimblegen SeqxCap EZ MedExome Target Enrichment Kit (Roche Sequencing, Pleasanton, CA, USA) followed by DNA sequencing by 74 base paired-end sequencing on the Illumina HiSeq 2000 instrument as previously described.17 Sequence metrics are shown in Supplementary Table 1. Sequence reads were mapped to the reference genome (HG38) with BWA-MEM18 and further processed using the GATK Best Practices workflows,18,19 which include duplication marking, indel realignment and base quality recalibration. Single nucleotide variants and small indels were called with GATK HaplotypeCaller and annotated using ANNOVAR,20 dbSNP (v138), 1000 Genomes (May 2013), NHLBI exome variant server and ExAC (v3).21
Kinship analysis
The relationship between the proband and parents was estimated using the pairwise identify-by-descent calculation in PLINK.22
De novo variant detection
De novo variants were called using the TrioDenovo program.23 TrioDenovo calculates the posterior likelihood of a mutation being a bona fide de novo event and assigned a data quality (DQ) score to each variant call. De novo candidates were filtered based on the following hard filters: (1) have a minor allele frequency (MAF) ⩽5×10−3 in ExAC, (2) pass GATK variant quality score recalibration, (3) have a minimum 10 reads total, 5 alternate allele reads and a 20% alternate allele ratio in proband, (4) have a minimum depth of 10 reference reads and alternate allele ratio <3% in parents (5) are exonic or canonical splice-site variants and (6) have a DQ⩾7 (suggested cutoff by authors of TrioDenovo). Finally, false positives were excluded by in silico visualization using Integrative Genomics Viewer (IGV)24 and BLAT search.
Dominant/recessive variant detection
Dominant variants were filtered for rareness (MAF⩽5×10−5 across 1000 Genomes, exome variant server or ExAC) and high-quality heterozygotes (pass GATK VQSR, have a minimum eight reads total, have a genotype quality score ⩾20, and have alternate allele ratio ⩾20%). The deleteriousness of missense mutations was predicted by the MetaSVM25 rank score (annotated as ’D-Mis’ if MetaSVM score ⩾0.83357). We filtered recessive variants for rare (MAF⩽10−3 across 1000 Genomes, exome variant server or ExAC) homozygous and compound heterozygous mutations that exhibited high-quality sequence reads (pass GATK VQSR, have a minimum eight reads total for both proband and parents, and have a genotype quality score ⩾20). Only loss-of-function mutations (nonsense, canonical splice-site and frameshift indels) and D-Mis were considered potentially damaging.
Targeted OCRL1 sequencing
The patient was enrolled at the Mayo Clinic’s Rare Kidney Stone Consortium and consent for molecular testing was obtained from the parents. Blood was drawn for DNA isolation and Sanger sequencing was performed on coding exons of OCRL1 (NM_000276) and flanking intronic regions using M13-tailed primers (Genewiz, South Plainfield, NJ, USA). Primer sequences are available on request. All Sanger chromatograms were analyzed using Mutation Surveyor, version 5.0.1 (Softgenics LLC, State College, PA, USA).
Results
The patient, an 11-year-old Caucasian male with a history of short stature, proteinuria, hypercalciuria and osteopenia, had undergone an extensive renal evaluation starting at the age of 5 that included an unremarkable renal ultrasound and an unremarkable renal biopsy. He has been treated with a medical regimen that included an angiotensin converting enzyme (ACE) inhibitor and a thiazide diuretic. A diagnosis of Dent’s disease was clinically suspected. He also had a history of occasional headaches and multiple food allergies and exhibited above-average intellect and excelled in school. His father and paternal grandfather have a history of kidney stones. However, his family history is negative for renal, neurological or ophthalmological disease. Notably, his mother is short in stature, with a medical history of ulcerative colitis and interstitial cystitis. She denied any renal symptoms, and did not present with abnormal serum electrolytes, creatinine or proteinuria on previous outpatient evaluation.
The neurosurgical consult service was contacted when the subject presented to the emergency room with low back pain 4 days after a minor fall in the sitting position from a height of ~3 feet while playing on his school’s jungle gym. On exam, the patient was normotensive (BP 96/71) and short in stature for his age (height 123 cm, <3rd percentile; weight 34.5 kg 43rd percentile). He was neurologically intact but exhibited point tenderness at his thoracic-lumbar junction. Radiographic evaluation including plain films and a computed tomography of his thoracolumbar spine revealed a T12 compression fracture with ~25% loss of vertebral body height and no significant canal compromise from retroplusion of bony fragments (Figure 1a). Magnetic resonance imaging of the entire spine showed no spinal cord compression from the fracture, but uncovered a significant Chiari I malformation (1.8 cm of tonsillar descent below the level of the foramen magnum; Figure 1b). This was associated with syringohydromyelia extending along the length of the cervical and thoracic cord, the largest diameter measuring ~3 mm at the level of C6 and C7. There was no evidence of hydrocephalus.

(a) Sagittal spine computed tomography image demonstrating a T12 compression fracture (arrow) that occurred after low-impact trauma in the setting of severe osteopenia. (b) Sagittal T2-weighted magnetic resonance image demonstrating significant (1.8 cm) herniation of the cerebellar tonsils (arrow) beyond the level of the foramen magnum, hallmark of a Chiari type 1 malformation. This was associated with syringohydromyelia extending through the length of the cervical and thoracic cord, the largest diameter of which measured ~3 mm at the level of C6 and C7.
Serum labs revealed normal levels of creatinine (0.58 mg/dl), ionized and total serum calcium, and phosphorus. Urine chemistries were remarkable for proteinuria (spot urine=0.86 g/l; protein/creatinine ratio=1.3 mg/mg), hypercalciuria (UCa/Cr ratio=0.38 mg/mg), elevated 1,25 dihydroxy vitamin D (73 pg/ml, normal=25–66 pg/ml), and normal 25 hydroxy vitamin D (37 ng/ml, normal=20–50 ng/ml). Urine β2 microglobulin was markedly elevated (90 mg/l, normal⩽0.23 mg/l). A renal ultrasound was unremarkable without evidence of kidney stones or nephrocalcinosis. A DEXA scan demonstrated severe osteopenia with a Z-score of −2.0. Given these findings, blood samples were collected from the child and his two biological parents. DNAs were isolated and sent for targeted sequencing of the genes known to cause Dent’s disease and whole-exome sequencing. The patient’s lumbar compression fracture was conservatively managed with a thoracolumbosacral orthosis (TLSO) brace. A suboccipital craniotomy with duroplasty and C1 laminectomy were electively performed for the Chairi I malformation months after his compression fracture had successfully healed.
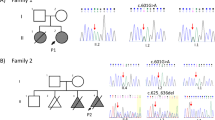
Screening of the proband demonstrated no mutations in the coding regions of CLCN5. Screening of OCRL1 by PCR and direct Sanger sequencing was performed as described in the Methods section. PCR failed to amplify exon 3 in the child compared with a positive control. Subsequently, PCR employing primers designed 101 bp 5′ of the Splice Acceptor Site of Exon 3 and 677 bp 3′ of the Splice Donor Site of Exon3 of the OCRL1 gene (available on request) revealed an abnormal fragment ~500 bp smaller when compared with those from both parents, providing evidence of a deletion within the genomic sequence in that region (Figure 2). Absence of the lower molecular weight fragment in the mother suggests this deletion is de novo. Targeted Sanger sequencing was then performed to further characterize the lower molecular weight product, which revealed a novel 462 bp deletion removing the final 13 nt of exon 3 and 449 nt of intron 3 (c187_199+449del; Figure 2). This deletion completely removes the exon 3 splice donor site, likely resulting in a fully penetrant splicing defect.

(a) Agarose gel electrophoresis of 1 kb Plus DNA ladder and PCR products amplified from the DNAs of affected child [K1_1], mother [K1_2] and father [K1_3]. PCR was performed using primers designed 101 bp 5′ of the Splice Acceptor Site of Exon 3 and 677 bp 3′ of the Splice Donor Site of Exon3 of the OCRL1 gene. Note the absence of the lower molecular weight fragment in the mother, suggesting de novo occurrence of the mutation in the affected child. (b) Schematic depicting the mutant sequence, harboring a novel, de novo 462 bp deletion: c.187_199+449del (p.Arg63fsX) involving the last 13 nt of exon 3 of OCRL1. The mutant sequence shows the frameshift in exon 3 and the out of frame amino acid sequence, assuming the deletion results in inclusion of the residual intron 3.
Suspecting the potential contribution of other genetic alterations given this patient’s atypical DD2 phenotype, whole-exome sequencing was performed on genomic DNA from the proband and both parents as described in the Methods section. The sequencing run achieved an average of 98.2 and 95.4% bases with at least 8x and 20x coverage, respectively (Supplementary Table 1). The identify-by-descent shared between the proband and parents is ~50% which confirms the parent–offspring relationship. Variants were called using the GATK Best Practices pipeline18,19 and annotated for protein change. The deleterious impact of missense variants was inferred using the MetaSVM algorithm.25 False positives were excluded by in silico visualization of aligned reads. Mutations that could contribute to significant results were validated by Sanger sequencing.
As parents did not have a history of renal involvement, it was suspected that the disease may be caused by either de novo or homozygous mutations in the context of recessive inheritance. To identify de novo mutations, the TrioDeNovo23 program was used and high-stringency filters were applied as described in the Methods section. We identified two de novo mutations in this trio. The first is a tolerated missense mutation in ZNF644 (p.L442F) and the second is a predicted-deleterious missense mutation in UBE2D4 (p.D112H; Supplementary Table 2). Both de novo mutations are absent among >105 alleles in ExAC. ZNF644, Zinc-Finger Protein 644, encodes a zinc-finger transcription factor, interacts with G9a to regulate gene expression during neurogenesis.26 In addition, mutations in ZNF644 have been associated with myopia.27 UBE2D4, Ubiquitin Conjugating Enzyme E2 D4, encodes a protein that is involved in the p53 and ubiquitination pathways.28 However, based on predicted functional impact and biological relevance, ZNF644 and UBE2D4 are unlikely to contribute to the phenotype. For the recessive genotypes, we did not identify any genes that harbor homozygotes or compound heterozygotes using an MAF cutoff of 10−3.
Next, we sought to identify rare (MAF⩽5×10−5) transmitted loss-of-function and D-Mis heterozygotes using filters described in the Methods section. In total, we identified 16 (7 loss-of-function and 9 D-Mis; Supplementary Table S3) transmitted protein-altering mutations. Among these, we focused our attention on a maternally inherited missense variant in INPP5B (c.152C>T; p.A51V; Figure 3a) in the affected child. The mutation was verified by Sanger sequencing of PCR products in the trio (Figure 3c). This variant is extremely rare, with an overall allele frequency of 8.4×10−6, with only 1 allele present in a total of 119,084 alleles in the ExAC database (http://exac.broadinstitute.org/about). Furthermore, no clinical implication of this single reported allele is present in the literature. INPP5B c.152C>T; p.A51V is completely conserved across 46 species analyzed (Figure 3b). This residue resides in the PH domain of INPP5B.13 In silico analyses of the impact of the mutation predicted by SIFT, PolyPhen2, MetaSVM and CADD algorithms were damaging/deleterious (Figure 3a; see Methods section). Nucleotide 152 is the last in exon 3, so we tested if the nucleotide substitution to T was predicted to alter splicing, however, analysis with the BDGP website predicted only a very slight weakening of splicing (0.95–0.94) that is not likely to be pathogenically significant.

(a) Analysis of the INPP5B c.152C>T; p.A51V variant. In silico analyses of mutation impact predicted by SIFT, PolyPhen2, MetaSVM and CADD algorithms are present. (b) Residue conservation analysis of the p.A51V mutation in INPP5B in orthologous proteins. The INPP5B p.A51V variant is shown with an arrow identifying the corresponding amino acid position. Protein sequences were downloaded from UniProt. The entries used for each species are as follows: P32019 (human), Q8K337 (mouse), D2A2N2 (rat), H2R981 (chimp), F1SV41 (pig), F76UML1 (horse) and F1NLk6 (chicken). (c) Family structure and Sanger sequence chromatograms from the trio with the INPP5B p.A51V mutation are shown. Affected individuals are denoted by filled squares while unaffected individuals are denoted by unfilled squares. The DNA sequence and the sequence of the encoded protein are shown in single letter code above sequence traces. Heterozygous mutations were detected in the patient and the mother.
Discussion
We report the first known case of an individual diagnosed with DD2 and Chiari I malformation and found to harbor mutations in the paralogs OCRL1 and INPP5B. Targeted sequencing revealed a novel, de novo deletion involving exon 3 of OCRL1, and whole-exome sequencing followed by Sanger sequencing revealed a heterozygous, non-synonymous missense variant in INPP5B. The latter is extremely rare based on ExAC, changes an evolutionarily conserved residue, resides in the protein’s functional PH domain, and is predicted to be highly deleterious. Mutations in INPP5B have not been previously associated with a human disease. In contrast, there have been ~20 cases of DD2 reported in the literature that result from loss-of-function mutations in the 5-phosphatase and PH domains of OCRL1. These OCRL1 mutations result in a spectrum of phenotypes that include low-molecular-weight proteinuria, hypercalciuria, nephrocalcinosis, hyperphosphaturia, aminoaciduria and intellectual disability.3,29–31 Structural brain malformations are notably absent in LS and DD2 associated with mutated OCRL1. We speculate that the combination of OCRL1 and INPP5B mutations in our patient is responsible for his unique clinical phenotype that includes a significant Chiari I malformation and syringohydromyelia in the cervical and thoracic cord.
Allelic differences in OCRL1 in Lowe syndrome and DD2 patients may explain the phenotypic variability between these two conditions. The majority of Lowe syndrome patients harbor nonsense or frameshift mutations in exons 9–22 of OCRL1, representing the region encoding the protein’s 5-phosphatase activity. Consistent with this finding, functional data demonstrate absence of the OCRL1 mRNA or a total or near-total lack of 5-phosphatase activity in Lowe syndrome patients.10,32 Conversely, DD2 patients harbor missense mutations in this same region of OCRL1, yielding protein products with reduced 5-phosphatase activity, or early truncating mutations in exons 1–7.3 It is worth noting that early truncating mutations in OCRL1 often produce clinically mild DD2. This has led some to hypothesize the existence of alternative isoforms of OCRL1 in the brains and eyes of DD2 patients as a possible explanation for the phenotypic differences between these two conditions.2
Furthermore, recent functional studies demonstrate accumulation of phosphatidylinositol 4,5-bisphosphate, abnormal F-actin and α-actin distribution, and impaired ciliogenesis in fibroblasts from both Lowe syndrome and DD2 patients. These changes were more drastic in Lowe syndrome fibroblasts, strengthening the hypothesis that DD2 may represent a milder form of Lowe sydrome.33
In vitro studies using human and animal cells have shown that the 5-phosphatase and RhoGAP domains are involved with the localization of ocrl1 to the primary cilium12,34 and its ability to modulate the length of the primary cilium.11 INPP5B has also been shown to be a positive modulator of ciliogenesis and ciliary length.35 It has been hypothesized that the essential role of OCRL1 in ciliogenesis may contribute to the pathology seen in DD2.33 The primary cilium is a non-motile projection on most eukaryotic cells, and links changes in the extracellular environment with intracellular signal transduction via a number of signaling pathways, including Sonic Hedgehog and possibly Wnt, key pathways in neurodevelopment.36,37 The primary cilium directly determines planar polarity of ependymal cells within the ventricles, and establishes the translational flow of cerebral spinal fluid.38 Investigators have speculated DD2 may be a part of a larger family of ’ciliopathies’, which are disorders frequently characterized by renal dysfunction, ataxia, cerebellar malformation and neurological deficits.39 Deficits in ciliogenesis and impaired cerebral spinal fluid flow have been associated with hydrocephalus40–42 and scoliosis43 which are found in increased frequency in patients with Chairi I syndrome.44 In the context of a DD2-causing OCRL1 mutation, we speculate that an INPP5B missense mutation may result in impaired ciliogenesis and contribute to Chairi I malformation and syringomyelia.
The genetic basis of Chiari I malformation is not well defined. Genome-wide association studies have identified genes involved in chondrogenesis, such as EP3000, CREBBP, SOX9, ATF4 and LHX4.45 A limited number of case reports have presented the association of this condition with abnormal skull morphology, especially due to a small posterior fossa in the context of bone disorders such as X-linked recessive hypophosphatemic rickets (OMIM #300554), or its X-linked dominant counterpart (OMIM # 307800).46,47 However, a clear mechanistic association between these conditions has not been accomplished. Further work is needed to define the genetics and mechanism of Chiari I malformation, and to examine further a possible pathogenic role of OCRL-dependent ciliogenesis in its pathogenesis.
References
Devuyst O, Thakker RV . Dent's disease. Orphanet J Rare Dis 2010; 5: 28.
Shrimpton AE, Hoopes RR Jr, Knohl SJ, Hueber P, Reed AA, Christie PT et al. OCRL1 mutations in Dent 2 patients suggest a mechanism for phenotypic variability. Nephron Physiol 2009; 112: p27–p36.
Hoopes RR Jr, Shrimpton AE, Knohl SJ, Hueber P, Hoppe B, Matyus J et al. Dent Disease with mutations in OCRL1. Am J Hum Genet 2005; 76: 260–267. Epub 2004 Dec 30. Erratum in: Am J Hum Genet 2007; 81(3):634.
Frymoyer PA, Scheinman SJ, Dunham PB, Jones DB, Hueber P, Schroeder ET et al. X-linked recessive nephrolithiasis with renal failure. N Engl J Med 1991; 325: 681–686.
Scheinman SJ . X-linked hypercalciuric nephrolithiasis: clinical syndromes and chloride channel mutations. Kidney Int 1998; 53: 3–17.
Gambaro G, Vezzoli G, Casari G, Rampoldi L, D'Angelo A, Borghi L . Genetics of hypercalciuria and calcium nephrolithiasis: from the rare monogenic to the common polygenic forms. Am J Kidney Dis 2004; 44: 963–986.
Tang X, Brown MR, Cogal AG, Gauvin D, Harris PC, Lieske JC et al. Functional and transport analyses of CLCN5 genetic changes identified in Dent disease patients. Physiol Rep 2016; 4: pii: e12776.
Bökenkamp A, Ludwig M . The oculocerebrorenal syndrome of Lowe: an update. Pediatr Nephrol 2016; 31: 2201–2212.
Lewis RA, Nussbaum RL, Brewer ED . Lowe Syndrome Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH (eds). University of Washington: Seattle, WA, USA, 2001 1993-2016.
Attree O, Olivos IM, Okabe I, Bailey LC, Nelson DL, Lewis RA et al. The Lowe's oculocerebrorenal syndrome gene encodes a protein highly homologous to inositol polyphosphate-5-phosphatase. Nature 1992; 358: 239–242.
Rbaibi Y, Cui S, Mo D, Carattino M, Rohatgi R, Satlin LM et al. OCRL1 modulates cilia length in renal epithelial cells. Traffic 2012; 13: 1295–1305.
Madhivanan K, Ramadesikan S, Aguilar RC . Role of Ocrl1 in primary cilia assembly. Int Rev Cell Mol Biol 2015; 317: 331–347.
Mao Y, Balkin DM, Zoncu R, Erdmann KS, Tomasini L, Hu F et al. A PH domain within OCRL bridges clathrin-mediated membrane trafficking to phosphoinositide metabolism. EMBO J 2009; 28: 1831–1842.
Jänne PA, Suchy SF, Bernard D, MacDonald M, Crawley J, Grinberg A et al. Functional overlap between murine Inpp5b and Ocrl1 may explain why deficiency of the murine ortholog for OCRL1 does not cause Lowe syndrome in mice. J Clin Invest 1998; 101: 2042–2053.
Bothwell SP, Chan E, Bernardini IM, Kuo YM, Gahl WA, Nussbaum RL et al. Mouse model for Lowe syndrome/Dent Disease 2 renal tubulopathy. J Am Soc Nephrol 2011; 22: 443–448.
Hellsten E, Evans JP, Bernard DJ, Jänne PA, Nussbaum RL . Disrupted sperm function and fertilin beta processing in mice deficient in the inositol polyphosphate 5-phosphatase Inpp5b. Dev Biol 2001; 240: 641–653.
Lemaire M, Fremeaux-Bacchi V, Schaefer F, Choi M, Tang WH, Le Quintrec M et al. Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat Genet 2013; 45: 531–536.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20: 1297–1303.
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 2013; 43: 11.10.1–11.10.33.
Wang K, Li M, Hakonarson H . ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010; 38: e164.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016; 536: 285–291.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Human Genet 2007; 81: 559–575.
Wei Q, Zhan X, Zhong X, Liu Y, Han Y, Chen W et al. A Bayesian framework for de novo mutation calling in parents-offspring trios. Bioinformatics 2015; 31: 1375–1381.
Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G et al. Integrative genomics viewer. Nat Biotechnol 2011; 29: 24–26.
Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet 2015; 24: 2125–2137.
Olsen JB, Wong L, Deimling S, Miles A, Guo H, Li Y et al. G9a and ZNF644 physically associate to suppress progenitor gene expression during neurogenesis. Stem Cell Rep 2016; 7: 454–470.
Shi Y, Li Y, Zhand D, Zhang H, Li Y, Lu F et al. Exome sequencing identifies ZNF644 mutations in high myopia. PLoS Genet 2011; 7: e1002084.
Lee JY, Tokumoto M, Fujiwara Y, Hasegawa T, Seko Y, Shimada A et al. Accumulation of p53 via down-regulation of UBE2D family genes is a critical pathway for cadmium-induced renal toxicity. Sci Rep 2016; 6: 21968.
Sekine T, Nozu K, Iyengar R, Fu XJ, Matsuo M, Tanaka R et al. OCRL1 mutations in patients with Dent disease phenotype in Japan. Pediatr Nephrol 2007; 22: 975–980.
Utsch B, Bökenkamp A, Benz MR, Besbas N, Dötsch J, Franke I et al. Novel OCRL1 mutations in patients with the phenotype of Dent disease. Am J Kidney Dis 2006; 48: 942.e1–14.
Hichri H, Rendu J, Monnier N, Coutton C, Dorseuil O, Poussou RV et al. From Lowe syndrome to Dent disease: correlations between mutations of the OCRL1 gene and clinical and biochemical phenotypes. Hum Mutat 2011; 32: 379–388.
Lin T, Orrison BM, Leahey AM, Suchy SF, Bernard DJ, Lewis RA et al. Spectrum of mutations in the OCRL1 gene in the Lowe oculocerebrorenal syndrome. Am J Hum Genet 1997; 60: 1384–1388.
Montjean R, Aoidi R, Desbois P, Rucci J, Trichet M, Salomon R et al. OCRL-mutated fibroblasts from patients with Dent-2 disease exhibit INPP5B-independent phenotypic variability relatively to Lowe syndrome cells. Hum Mol Genet 2015; 24: 994–1006.
Luo N, West CC, Murga-Zamalloa CA, Sun L, Anderson RM, Wells CD et al. OCRL localizes to the primary cilium: a new role for cilia in Lowe syndrome. Hum Mol Genet 2012; 21: 3333–3344.
Kim J, Lee JE, Heynen-Genel S, Suyama E, Ono K, Lee K et al. Functional genomic screen for modulators of ciliogenesis and cilium length. Nature 2010; 464: 1048–1051.
Sattar S, Gleeson JG . The ciliopathies in neuronal development: a clinical approach to investigation of Joubert syndrome and Joubert syndrome-related disorders. Dev Med Child Neurol 2011; 53: 793–798.
Wallingford JB . Planar cell polarity, ciliogenesis and neural tube defects. Hum Mol Genet 2006; 15: R227–R234.
Mirzadeh Z, Han YG, Soriano-Navarro M, García-Verdugo JM, Alvarez-Buylla A . Cilia organize ependymal planar polarity. J Neurosci 2010; 30: 2600–2610.
Schurman SJ, Scheinman SJ . Inherited cerebrorenal syndromes. Nat Rev Nephrol 2009; 5: 529–538.
Lechtreck KF, Delmotte P, Robinson ML, Sanderson MJ, Witman GB . Mutations in Hydin impair ciliary motility in mice. J Cell Biol 2008; 180: 633–643.
Ibañez-Tallon I, Gorokhova S, Heintz N . Loss of function of axonemal dynein Mdnah5 causes primary ciliary dyskinesia and hydrocephalus. Hum Mol Genet 2002; 11: 715–721.
Banizs B, Pike MM, Millican CL, Ferguson WB, Komlosi P, Sheetz J et al. Dysfunctional cilia lead to altered ependyma and choroid plexus function, and result in the formation of hydrocephalus. Development 2005; 132: 5329–5339.
Cheng JC, Castelein RM, Chu WC, Danielsson AJ, Dobbs MB, Grivas TB et al. Adolescent idiopathic scoliosis. Nat Rev Dis Primers 2015; 1: 15030.
Milhorat TH, Chou MW, Trinidad EM, Kula RW, Mandell M, Wolpert C et al. Chiari I malformation redefined: clinical and radiographic findings for 364 symptomatic patients. Neurosurgery 1999; 44: 1005–1017.
Markunas CA, Lock E, Soldano K, Cope H, Ding CK, Enterline DS et al. Identification of Chiari Type I Malformation subtypes using whole genome expression profiles and cranial base morphometrics. BMC Med Genomics 2014; 7: 39.
Watts L, Wordsworth P . Chiari malformation, syringomyelia and bulbar palsy in X linked hypophosphataemia. BMJ Case Rep 2015; 2015: pii: bcr2015211961.
Caldemeyer KS, Boaz JC, Wappner RS, Moran CC, Smith RR, Quets JP . Chiari I malformation: Association with hyophosphatemic rickets and MR imaging appearance. Radiology 1995; 195: 733–738.
Acknowledgements
This study was supported by the Rare Kidney Stone Consortium (U54KD083908), a part of the Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR), National Center for Advancing Translational Sciences (NCATS). This consortium is funded through a collaboration between NCATS, and National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). The study sponsor had no role in study design; collection, analysis and interpretation of data; writing the report; and the decision to submit the report for publication. We thank Barb Seide for excellent study coordination. This work was also supported by NIH Medical Scientist Training Program Training Grant (T32GM007205). Dr Kahle is supported by March of Dimes, The Simons foundation and the NIH—Centers for Mendelian Genomics.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information for this article can be found on the Human Genome Variation website .
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Duran, D., Jin, S., DeSpenza, T. et al. Digenic mutations of human OCRL paralogs in Dent’s disease type 2 associated with Chiari I malformation. Hum Genome Var 3, 16042 (2016). https://doi.org/10.1038/hgv.2016.42
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2016.42
This article is cited by
-
Development of ultra-deep targeted RNA sequencing for analyzing X-chromosome inactivation in female Dent disease
Journal of Human Genetics (2018)
