
Abstract
Distal arthrogryposis (DA) is a clinically and genetically heterogeneous disorder with multiple joint contractures. We describe a female DA patient with hand and foot deformities, and right-sided torticollis. Using exome sequencing, we identified a novel TNNI2 mutation (c.485>A, p.Arg162Lys) in the patient and her father. The father has no typical DA but hip dysplasia. This may explain the clinical features of DA2B in this family, but with variable clinical expression.
Similar content being viewed by others
Distal arthrogryposis (DA) is a clinically and genetically heterogeneous disorder characterized by congenital limb contractures with no primary neurological or muscular effects. DA is an autosomal dominant disorder with reduced penetrance and variable expression. DA is classified into 10 types, 1, 2A, 2B, 3–5, 7–10,1 and molecular diagnosis can help with the correct diagnosis of DA. DA2B (OMIM 601680) is one of the most common types.2 In DA2B, mutations have been found in genes encoding troponin I, fast skeletal type (TNNI2),3 troponin T3, fast skeletal type (TNNT3),4 myosin heavy chain 3, skeletal muscle, embryonic (MYH3)5 and tropomyosin 2 (TPM2).5 In this study, we describe the detailed clinical features and molecular diagnosis of a female DA patient.
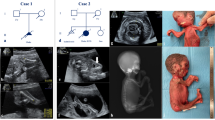
The female patient was 3 years old and born after 40 weeks of gestation to non-consanguineous Croatian parents (Figure 1a). Apgar scores were 8/7 at 1/5 min and her birth weight was 3450 g (50th percentile) and she was 48 cm long (25th percentile). At birth, short neck, low posterior hairline, nevus flammeus on the forehead and asymmetric epicanthus were seen. Bilateral joint contractures were also observed at the wrists, elbows, hips and knees, together with right-sided torticollis. In addition, hand and foot deformities (talipes equinovarus) and skin-furrowed fingers were reported (Table 1). Serum amino acid and organic acid levels, peripheral blood lymphocyte karyotypes and electroencephalogram were all normal. During the first few months, the patient showed motor delay, bilateral hypertonia and vertical talus (Figure 1b, c). Her motor movements were slow, and closed fists and bilateral unabducted thumbs were observed (Figure 1d, e). Her head was tilted to the left and rotated to the right. The patient also presented with hypertonic upper limbs and a palmar grasp. In addition, she was unstable when standing on tiptoes. Physiological reflexes were present. Physical therapy was started immediately for neurodevelopmental delay and right-sided torticollis. Poor muscle tone, hypertonus of the upper limbs, and palmar grasp were consistently observed. Conservative orthopedic correction using the Dobbs method (high gypsum boots, replaced every 7 days) was started. At 4 months, the talus was surgically repositioned and the navicular bone was fixed with Kirschner wire. The patient wore orthoses for her ankles and feet during the day, but only at night on the hands and wrists. Physical therapy is currently ongoing.

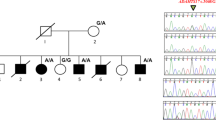
(a) Pedigree tree and segregation status of TNNI2 mutation (c.485G>A, p.Arg162Lys). WT: wild type, Mut: mutant (b–e) The female patient showing vertical talus (b, c) and overlapping fingers (d, e). Electropherograms around the mutation of the patient and her parents (f). The father had hip dysplasia (g). TNNI2 mutations were mapped to the protein (h). Functional domains are colored differently (ID: inhibitory region, SR: switch region). The current (red) and reported (black) mutations were clustered in the actin-binding domain (h). Schematic representation of the troponin complex with an actin polymer in low Ca2+ concentrations, modified from Murakami et al. (i).13 The Ca2+-binding (TnC) and tropomyosin-binding (TnT) subunits are depicted as a yellow dumbbell shape and a magenta tube, respectively. The domain organization of the inhibitory subunit (TnI)14 is indicated; the IT-arm, which comprises two helices connected by a flexible linker, the inhibitory region, the switch region and the C-terminal actin-binding domain, is shown as cyan tube-like shapes connected with a cyan string, a dark-blue string, a pink rectangle and string, and a green oval, respectively. Actin monomers are presented as colored rounded rectangles. The mutation position of human TnI was mapped in a solution of the actin-binding domain of chicken TnI in a Ca2+-free state (PDB code 1VDI).13 The mutation site is colored red. The side chains of the residue at the mutation site (Arg161 in chicken) and its surrounding residues are shown as van der Waals spheres. The amino acid number of chicken TnI is shown with that of human TnI in parentheses. N and C denote the N- and C-terminus of the protein.
Whole-exome sequencing (WES) was performed as previously described6 and 93.7% of RefSeq coding regions were covered by 20 or more reads. WES identified a novel TNNI2 mutation (NM_003282, c.485G>A, p.Arg162Lys) in the patient and her father (Figure 1a, f). The altered amino residue is evolutionally conserved from Xenopus tropicalis to Homo sapiens. This missense mutation was predicted to be pathogenic by three prediction software: SIFT: damaging (score=0), PolyPhen-2: possibly damaging (score=0.956) and MutationTaster: disease causing. This variant has not been reported in the available public databases including ExAC browser (http://exac.broadinstitute.org/), Exome Variant Server (http://evs.gs.washington.edu/EVS/) or 1000 Genomes (http://browser.1000genomes.org/index.html) (all accessed on July 29, 2016). Sanger sequencing revealed that this mutation occurred de novo in the proband’s father (Figure 1a). The father did not have the clinical symptoms of DA, but he did have hip dysplasia in infancy, which was surgically repaired (Figure 1g). A total of 10 TNNI2 mutations have been reported.2,3,5,7–10 All TNNI2 mutations, including the mutation reported here, occur in a small region (156–178 amino acids (aa)) of the TNNI2 actin-binding domain (182 aa in length) (Figure 1h). Therefore, this small region probably has an important role in TNNI2 function.
The troponin complex consists of a Ca2+-binding subunit (TnC), a tropomyosin-binding subunit (TnT) and an inhibitory subunit (TnI) (Figure 1i). Mutations are specific to the TnI subunit and exclusively affect the fast skeletal muscle. Low Ca2+ concentrations stabilize the inhibitory conformation of the troponin-tropomyosin complex, which inhibits actomyosin interactions, leading to relaxation of the skeletal muscle.11 The C-terminal actin-binding domain of TnI is required for Ca2+-dependent inhibition12 (Figure 1i).
We mapped the position of the human TnI mutation using the actin-binding domain of chicken TnI in a Ca2+-free state (PDB code 1VDI).13 The mutation occurred in a conserved arginine residue (Arg162 in human and Arg161 in chicken) located within the actin-binding domain of TnI. As shown in Figure 1i, a methylene group of the arginine is a component of the hydrophobic core and this can be replaced by the methylene group of the mutated lysine residue (p.Arg162Lys). Therefore, this mutation does not affect the protein structure, but could impair the molecular interactions between TnI and actin filaments.
In conclusion, we describe a novel TNNI2 mutation in two family members: a young girl with DA2B and her father with hip dysplasia (but not typical DA). This mutation may explain the variable clinical expression of DA2B in this family.
References
References
Bamshad M, Van Heest AE, Pleasure D . Arthrogryposis: a review and update. J Bone Joint Surg Am 2009; 91: 40–46.
Wang B, Zheng Z, Wang Z, Zhang X, Yang H, Cai H et al. A novel missense mutation of TNNI2 in a Chinese family cause distal arthrogryposis type 1. Am J Med Genet A 2016; 170: 135–141.
Sung SS, Brassington AM, Grannatt K, Rutherford A, Whitby FG, Krakowiak PA et al. Mutations in genes encoding fast-twitch contractile proteins cause distal arthrogryposis syndromes. Am J Hum Genet 2003; 72: 681–690.
Sung SS, Brassington AM, Krakowiak PA, Carey JC, Jorde LB, Bamshad M . Mutations in TNNT3 cause multiple congenital contractures: a second locus for distal arthrogryposis type 2B. Am J Hum Genet 2003; 73: 212–214.
Beck AE, McMillin MJ, Gildersleeve HI, Kezele PR, Shively KM, Carey JC et al. Spectrum of mutations that cause distal arthrogryposis types 1 and 2B. Am J Med Genet A 2013; 161A: 550–555.
Nakashima M, Takano K, Tsuyusaki Y, Yoshitomi S, Shimono M, Aoki Y et al. WDR45 mutations in three male patients with West syndrome. J Hum Genet 2016; 61: 653–661.
Jiang M, Zhao X, Han W, Bian C, Li X, Wang G et al. A novel deletion in TNNI2 causes distal arthrogryposis in a large Chinese family with marked variability of expression. Hum Genet 2006; 120: 238–242.
Carli D, Fairplay T, Ferrari P, Sartini S, Lando M, Garagnani L et al. Genetic basis of congenital upper limb anomalies: analysis of 487 cases of a specialized clinic. Birth Defects Res A Clin Mol Teratol 2013; 97: 798–805.
Shrimpton AE, Hoo JJ . A TNNI2 mutation in a family with distal arthrogryposis type 2B. Eur J Med Genet 2006; 49: 201–206.
Kimber E, Tajsharghi H, Kroksmark AK, Oldfors A, Tulinius M . Distal arthrogryposis: clinical and genetic findings. Acta Paediatr 2012; 101: 877–887.
Parry DA, Squire JM . Structural role of tropomyosin in muscle regulation: analysis of the X-ray diffraction patterns from relaxed and contracting muscles. J Mol Biol 1973; 75: 33–55.
Rarick HM, Tu XH, Solaro RJ, Martin AF . The C terminus of cardiac troponin I is essential for full inhibitory activity and Ca2+ sensitivity of rat myofibrils. J Biol Chem 1997; 272: 26887–26892.
Murakami K, Yumoto F, Ohki SY, Yasunaga T, Tanokura M, Wakabayashi T . Structural basis for Ca2+-regulated muscle relaxation at interaction sites of troponin with actin and tropomyosin. J Mol Biol 2005; 352: 178–201.
Takeda S, Yamashita A, Maeda K, Maeda Y . Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form. Nature 2003; 424: 35–41.
Data Citations
Matsumoto, Naomichi HGV Database (2016) http://dx.doi.org/10.6084/m9.figshare.hgv.882
Acknowledgements
We thank the patient and her family for their participation in this study. This work was supported by grants from Research on Measures for Intractable Diseases; Comprehensive Research on Disability Health and Welfare; the Strategic Research Program for Brain Science, Practical Research Project for Rare/Intractable Diseases, the Initiative on Rare and Undiagnosed Diseases in Pediatrics and for Adults from the Japan Agency for Medical Research and Development; a grant-in-aid for Scientific Research on Innovative Areas (Transcription Cycle) from the Ministry of Education, Culture, Sports, Science and Technology of Japan; grants-in-aid for Scientific B and C from the Japan Society for the Promotion of Science; the fund for Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems from the Japan Science and Technology Agency; and the Takeda Science Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Čulić, V., Miyake, N., Janković, S. et al. Distal arthrogryposis with variable clinical expression caused by TNNI2 mutation. Hum Genome Var 3, 16035 (2016). https://doi.org/10.1038/hgv.2016.35
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2016.35
This article is cited by
-
Distal arthrogryposis in a girl arising from a novel TNNI2 variant inherited from paternal somatic mosaicism
Journal of Human Genetics (2023)
-
Rare clinical phenotype of filaminopathy presenting as restrictive cardiomyopathy and myopathy in childhood
Orphanet Journal of Rare Diseases (2022)
