
Abstract
Simpson–Golabi–Behmel syndrome is a congenital malformation syndrome associated with mutations in GPC3, which is located in the Xq26 region. Three new loss-of-function mutations and a global X-chromosome rearrangement involving GPC3 were identified. A female sibling of the patient, who presented with a cleft palate and hepatoblastoma, carries the same chromosomal rearrangement and a paradoxical pattern of X-chromosome inactivation. These findings support variable GPC3 alterations, with a possible mechanism in female patients.
Similar content being viewed by others


Simpson–Golabi–Behmel syndrome type 1 (SGBS1; MIM #312870) is a congenital malformation syndrome characterized by pre/postnatal overgrowth, intellectual disability and distinctive craniofacial features including macrocephaly, coarse facial features, macrostomia, macroglossia and palatal abnormalities.1,2 Diagnosis of SGBS is based on the clinical findings, family history consistent with X-linked inheritance and molecular biological testing of the glypican-3 gene (GPC3; MIM #300037 [http://omim.org/]) that maps to Xq26. GPC3, a member of the glypican family, encodes a heparan sulfate proteoglycan that is attached to the cell membrane via a glycosylphosphatidylinositol linkage.3 The gene spans >500 kb of genomic DNA, contains eight exons, and has essential roles in development by modulating cellular responses to growth factors and morphogens. Here, we report the identification of novel GPC3 mutations: three nucleotide alterations and one global chromosomal rearrangement involving GPC3.
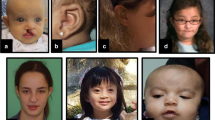
Patient 1 was a 7-year-old boy with non-consanguineous parents, born at 26 weeks of gestation by emergency cesarean section due to fetal distress. His birth weight, length and occipitofrontal circumference (OFC) were 976 g (>90th centile), 37 cm (75–90th centile) and 24.5 cm (50th centile), respectively, which were considered large for his gestational age. The Apgar scores were 1 and 3 at 1 and 5 min, respectively. He presented with generalized hypotonia and required tracheal intubation for 8 days. Because he had difficulty in feeding owing to poor sucking in early infancy, gastric tube feeding was employed. He exhibited global developmental delay from the neonatal period and started walking alone at 30 months. His developmental quotient was 29, as determined by the Enjoji developmental test at 20 months. The child had hypothyroidism, febrile convulsions, ventricular septal defect, sleep apnea syndrome induced by an enlarged tonsil that needed to be removed and repeated otitis media with effusion. At 5 years, his height, weight and OFC were 114 cm (>90th centile), 23 kg (>90th centile) and 52.5 cm (50–75th centile), respectively, indicating postnatal overgrowth. He exhibited ocular hypertelorism, epicanthal folds, upslanting palpebral fissures, a wide nasal bridge, macrostomia (an abnormally large mouth), macroglossia (an abnormally large tongue), dental malocclusion, accessory nipples, broad fingers, bilateral planovalgus and genu valgum.
Patient 2 was a 3-month-old male infant born at 32 weeks by cesarean section because of threat of premature labor. His birth weight was 2,560 g and his length was 45 cm, indicating over 90th centile for both. Both his parents were 23 years old and healthy; their first child was also healthy. Before birth, congenital diaphragmatic hernia was noted, and the hernia was surgically repaired at 5 days after birth. Physical examination showed a large tongue and big toes. Abdominal echography showed that both kidneys were on the left side.
Patient 3 was a 10-year-old boy born at 39 weeks of gestation. His birth weight, length and OFC were 4,030 g (>97th centile), 55 cm (>97th centile) and 36 cm (>97th centile), respectively, indicating prenatal overgrowth. Fetal ultrasonography suggested congenital diaphragmatic hernia, which was surgically repaired soon after birth. Ileus due to midgut volvulus was noted at 3 months and partial resection of the intestine was required. At present, his height, weight and OFC were 141.9 cm (75–90th centile), 29.8 kg (25–50th centile) and 52.0 cm (10–25th centile), respectively. He exhibited coarse facial features with macroglossia, and bilateral accessory nipples were noted. His developmental milestones were within the normal limit.
Patient 4 was an 18-month-old boy born at 38 weeks of gestation. His birth weight, length and OFC were 4,040 g (>97th centile), 55 cm (>97th centile) and 36 cm (>97th centile), respectively, indicating prenatal overgrowth. He was the second child of a mother who had a cleft palate at birth. The patient presented some dysmorphic features, such as macrocephaly, macroglossia, a cleft palate, hydronephrosis, bilateral accessory nipples and a prostatic utricle. He started walking at 18 months. At present, his height, weight and OFC were 85.0 cm (90–97th centile), 12.9 kg (>97th centile) and 49.0 cm (75–90th centile), respectively.
Patient 5 was a 3-year-old girl (the elder sister of patient 4) born at 37 weeks of gestation. Her birth weight, length and OFC were 3,600 g (>97th centile), 52 cm (>97th centile) and 35 cm (90–97th centile), respectively. She also had a cleft palate, which was surgically repaired at 12 months. At 19 months, she was examined for hepatomegaly and found to have a highly elevated alpha fetoprotein level (140,000 ng/ml). Medical examination identified a hepatoblastoma, which was surgically resected soon after. At present, her height, weight and OFC were 100.3 cm (>97th centile), 16.3 kg (90–97th centile) and 50.3 cm (90th centile), respectively. Her development was within the normal limits.
On the basis of the clinical diagnosis of SGBS, genetic analyses were performed for the patients as described.4 This study was approved by the ethics committee of the institutions. After obtaining written informed consent from the patients’ families, peripheral blood samples were collected from the patients and genomic DNA was extracted using the QIAamp DNA extraction kit (QIAgen, Hilden, Germany). Using standard Sanger sequencing, we identified mutations in three of the patients (Supplementary Figure S1): patient 1—NM_004484(GPC3_v001):c.90_91dup [NM_004484(GPC3_i001): p.(Pro31Argfs*54)]; patient 2—c.758del [p.(Gly253Alafs*16)]; patient 3—c.1330C>T [p.(Gln444*)]. All mutations are predicated to be loss-of-function mutations. The mother of patient 1 was shown to harbor no mutation, suggesting a de novo occurrence. The parents of patients 2 and 3 declined genotyping of their DNA.
Because no nucleotide change was identified in patient 4, whole-genome copy-number analysis was performed using the Agilent 60 K Human Genome CGH Microarray platform (Agilent Technologies, Santa Clara, CA, USA), as described previously.5 Two microduplications were identified on the X chromosome, one of which was in GPC3 (Supplementary Figure S2). To evaluate the details of these microduplications, a custom array was designed and used, as described previously.6 The final molecular karyotype was arr Xq26.2 (132,742,336−132,933,597)×2,Xq26.3(135,873,188−136,230,169)×2 (GRCh37/hg19) (Figure 1a), indicating microduplication sizes of 191 and 357 kb. The microduplication in Xq26.2 spans exons 3–6 of GPC3. Patient 5 also harbors two microduplications, which suggests that the two microduplications segregate together. Thus, we suspected the existence of a complicated chromosomal rearrangement involving two regions. To test this hypothesis, we attempted to genotype chromosomal breakpoints and fused regions utilizing various sets of primers. Only one amplicon was successfully generated using the following primer set: primer 1, 5′- GCAACCTCAGTTCCCTGAAG-3′, and primer 2, 5′- GACAGAGCCTTGCTCTGCAACC-3′ (Figure 1b). Sequence analysis of this 1,070 bp amplicon included the breakpoints of both microduplications, indicating that the microduplications were fused by a complex chromosomal rearrangement, with an insertion of 16 bp of the unknown sequence (Figure 1c). PCR amplicons were also generated using DNA from patient 5 and the mother, as well as a healthy, maternal aunt of patients 4 and 5. All amplicons were the same size, indicating that the aunt is also a non-symptomatic carrier (Figure 1b).

Molecular analysis of DNA from patient 4. (a) Custom array analysis using Gene View created by Agilent Genomic Workbench (Agilent Technologies, Santa Clara, CA, USA) demonstrates two microduplications in the regions of Xq26.2 (up) and Xq26.3 (bottom). Two primers (blue arrows) were designed based on the microduplications. (b) Electropherogram of the 1,070 b.p. amplicon (black arrow) detected in patients 4 and 5, their mother and their aunt. C, A normal control sample. M, marker of φX174/HaeIII digest. (c) The result of Sanger sequence of the 1070-bp amplicon. Between two breakpoints, a 16-bp unknown sequence is inserted.
In general, SGBS is inherited as an X-linked recessive trait; however, patient 5 (female) displayed partial SGBS symptoms (a cleft palate and hepatoblastoma). Thus, the X-chromosome inactivation pattern was analyzed by the human androgen receptor gene assay, as described previously.7 The mother of patients 4 and 5 showed a skewed X-chromosome inactivation pattern (82% vs. 18%), which was in agreement with her lack of symptoms (Figure 2). In contrast, patient 5 showed a paradoxical X-chromosome inactivation pattern, with the affected allele being predominantly activated.

X-chromosome inactivation (XCI) study for obligate carriers. The shorter allele is common in patient 4 and indicates the affected allele (red rectangles). HpaII digestion of the non-methylated allele; the longer allele is predominantly digested in the mother’s sample (82% vs 18%), indicating skewed XCI (an asterisk; XCI is defined as skewed when the ratio of peaks differs >80:20). In contrast, patient 5 showed a paradoxical XCI pattern, with the affected allele (two asterisks) being predominantly inactivated.
More than 56 GPC3 mutations have been reported, with most being loss of function,4,8,9 which is consistent with the findings of the current study because all three identified mutations are predicted to be nonsense or frameshift mutations. The most intriguing finding was a microduplication involving GPC3 exons 3–6. Two intragenic duplications have been reported.10,11 In comparison with those duplications, the microduplication identified in this study is complicated, originating from a distal Xq26.3 region. Furthermore, the mother of patient 4 and patient 5 (the elder sister) had a cleft palate, which is one of the findings of SGBS. Patient 5 also had a hepatoblastoma. Phenotypic differences between female carriers appear to depend on the X-chromosome inactivation pattern.12
References
References
Cottereau E, Mortemousque I, Moizard MP, Burglen L, Lacombe D, Gilbert-Dussardier B et al. Phenotypic spectrum of Simpson-Golabi-Behmel syndrome in a series of 42 cases with a mutation in GPC3 and review of the literature. Am J Med Genet C Semin Med Genet 2013; 163C: 92–105.
Tenorio J, Arias P, Martinez-Glez V, Santos F, Garcia-Minaur S, Nevado J et al. Simpson-Golabi-Behmel syndrome types I and II. Orphanet J Rare Dis 2014; 9: 138.
Pilia G, Hughes-Benzie RM, MacKenzie A, Baybayan P, Chen EY, Huber R et al. Mutations in GPC3, a glypican gene, cause the Simpson-Golabi-Behmel overgrowth syndrome. Nat Genet 1996; 12: 241–247.
Sakazume S, Okamoto N, Yamamoto T, Kurosawa K, Numabe H, Ohashi Y et al. GPC3 mutations in seven patients with Simpson-Golabi-Behmel syndrome. Am J Med Genet A 2007; 143A: 1703–1707.
Sangu N, Okamoto N, Shimojima K, Ondo Y, Nishikawa M, Yamamoto T . A de novo microdeletion in a patient with inner ear abnormalities suggests that the 10q26.13 region contains the responsible gene. Hum Genome Var 2016; 3: 16008.
Shimojima K, Mano T, Kashiwagi M, Tanabe T, Sugawara M, Okamoto N et al. Pelizaeus-Merzbacher disease caused by a duplication-inverted triplication-duplication in chromosomal segments including the PLP1 region. Eur J Med Genet 2012; 55: 400–403.
Shimada S, Okamoto N, Ito M, Arai Y, Momosaki K, Togawa M et al. MECP2 duplication syndrome in both genders. Brain Dev 2013; 35: 411–419.
Okamoto N, Yagi M, Imura K, Wada Y . A clinical and molecular study of a patient with Simpson-Golabi-Behmel syndrome. J Hum Genet 1999; 44: 327–329.
Veugelers M, Cat BD, Muyldermans SY, Reekmans G, Delande N, Frints S et al. Mutational analysis of the GPC3/GPC4 glypican gene cluster on Xq26 in patients with Simpson-Golabi-Behmel syndrome: identification of loss-of-function mutations in the GPC3 gene. Hum Mol Genet 2000; 9: 1321–1328.
Mateos ME, Beyer K, Lopez-Laso E, Siles JL, Perez-Navero JL, Pena MJ et al. Simpson-Golabi-Behmel syndrome type 1 and hepatoblastoma in a patient with a novel exon 2-4 duplication of the GPC3 gene. Am J Med Genet A 2013; 161A: 1091–1095.
Cottereau E, Moizard MP, David A, Raynaud M, Marmin N, Toutain A . Duplication of exon 2 of the GPC3 gene in a case of Simpson-Golabi-Behmel syndrome. Am J Med Genet A 2014; 164A: 282–284.
Yano S, Baskin B, Bagheri A, Watanabe Y, Moseley K, Nishimura A et al. Familial Simpson-Golabi-Behmel syndrome: studies of X-chromosome inactivation and clinical phenotypes in two female individuals with GPC3 mutations. Clin Genet 2011; 80: 466–471.
Data Citations
Yamamoto, Toshiyuki HGV Database (2016) http://dx.doi.org/10.6084/m9.figshare.hgv.858
Yamamoto, Toshiyuki HGV Database (2016) http://dx.doi.org/10.6084/m9.figshare.hgv.861
Yamamoto, Toshiyuki HGV Database (2016) http://dx.doi.org/10.6084/m9.figshare.hgv.864
Yamamoto, Toshiyuki HGV Database (2016) http://dx.doi.org/10.6084/m9.figshare.hgv.867
Yamamoto, Toshiyuki HGV Database (2016) http://dx.doi.org/10.6084/m9.figshare.hgv.870
Acknowledgements
We would like to express our gratitude to the patients and their parents for their cooperation. This research was supported by the Practical Research Project for Rare/Intractable Diseases from the Japan Agency for Medical Research and Development (AMED) and the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Number 15K09631 (TY).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information for this article can be found on the Human Genome Variation website (http://www.nature.com/hgv)
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Shimojima, K., Ondo, Y., Nishi, E. et al. Loss-of-function mutations and global rearrangements in GPC3 in patients with Simpson–Golabi–Behmel syndrome. Hum Genome Var 3, 16033 (2016). https://doi.org/10.1038/hgv.2016.33
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2016.33
This article is cited by
-
A novel quantitative targeted analysis of X-chromosome inactivation (XCI) using nanopore sequencing
Scientific Reports (2023)
