
Abstract
Mutations in KIF7, the gene that encodes a component of the kinesin complex of anterograde intraflagellar transport in the cilia, have been reported to cause a range of phenotypes including hydrolethalis, acrocallosal syndrome and Joubert syndrome. In a cohort of patients with various neurogenetic phenotypes, we identified novel KIF7 mutations in two families that span the known phenotypic spectrum of KIF7-related disorders. Surprisingly, we also identified a novel truncating KIF7 mutation in a third consanguineous family, in which the index presented with intellectual disability but no overt signs of ciliopathy, and his brain magnetic resonance imaging revealed an isolated dysgenesis of corpus callosum. This small cohort contributes novel pathogenic alleles of KIF7 and suggests that KIF7-related phenotypes can include isolated dysgenesis of corpus callosum with intellectual disability, thus expanding the range of phenotypes that warrant sequencing of this gene.
Similar content being viewed by others
Ciliopathies are clinical disorders that trace their pathogenesis to the defects in the primary cilium, a microtubule-based antenna-like cellular organelle that is implicated in a wide array of cellular processes.1 This common pathoetiology underpins the remarkable phenotypic overlap between various ciliopathies that were once considered distinct clinical entities, and readily explains some of the recurrent phenotypic features such as retinal degeneration (caused by loss of the connecting cilium in the photoreceptors), polydactyly (caused by abnormal ciliary signaling response to the Sonic Hedgehog morphogen gradient) and cystic kidney disease (caused by abnormal ciliary mechanical response to the urine flow).2
Central nervous system involvement in ciliopathies is complex and ranges from the very mild or absent to the profound and lethal malformations with predilection to the posterior fossa as often observed in Meckel–Gruber syndrome.3 Acrocallosal syndrome (ACS) is a ciliopathy with a prominent central nervous system involvement usually in the form of absent corpus callosum and hydrocephalus in addition to polydactyly. KIF7 (Kinesin Family Member Protein 7) mutations have been found to cause a spectrum of phenotypes that collectively fall under the clinical definition of ACS.4 In this report, we emphasize the marked phenotypic variability of KIF7-linked phenotypes ranging from the perinatally lethal to a previously unrecognized mild end of the spectrum that is limited to intellectual disability and absence of corpus callosum.
We report three cases with KIF7 mutation, one with a classical ACS diagnosis. Another case had brain features consistent with the diagnosis of Joubert Syndrome, and KIF7 sequencing revealed a pathogenic mutation making it among the veryfew reported Joubert cases to carry a mutation in this gene. The third case was very mild and the phenotype was one of intellectual disability and dysgenesis of corpus callosum dysgenesis but no other signs of ciliopathy.
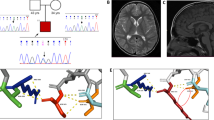
Case 1 is a 9-months-old boy born to consanguineous parents. Birth weight was 3.2 kg and head circumference was unknown. He started having recurrent seizures at 4 months of age. Evaluation at 9 months showed global developmental delay, dysmorphic facial features, bilateral pre-axial lower limb polydactyl and hypotonia (Figure 1). Brain computarized tomography revealed corpus callosum agenesis. Case 2 is a 2.5-years-old boy with a global developmental delay. Magnetic resonance imaging revealed a classical molar tooth sign, inferior vermis hypoplasia and subsequent secondary changes in the posterior fossa and corpus callosum agenesis (Figure 1). Case 3 is a 4 year-old boy born to consanguineous parents. Delivery was complicated by perinatal asphyxia necessitating neonatal intensive care unit management. Evaluation at four years of age revealed facial dysmorphism with brachycephaly, frontal bossing, flat supraorbital ridges, prognathism, everted lower lip and saddle nose with a tuft of hair surrounding a sinus. Two café au lait spots were noted. Magnetic resonance imaging-brain revealed mild dysgenesis of the corpus callosum (Figure 1).

Family pedigrees and clinical pictures of the three reported cases. Case 1: note the dysmorphic facial features and bilateral pre-axial lower limb polydactyl. Magnetic resonance imaging shows a classical molar tooth sign, inferior vermis hypoplasia and subsequent secondary changes in the posterior fossa and corpus callosum agenesis. Case 3: note the overlapping facial features with Case 1; magnetic resonance imaging-brain reveals dysgenesis of the corpus callosum.
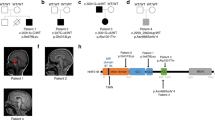
Patients and available family members were recruited using an institutional review board approved protocol (KFSHRC RAC# 2080006) and a written informed consent was obtained from all subjects (or legal guardians) prior to enrollment. Venous blood was collected in EDTA tubes for DNA extraction and molecular studies. In Case 1 where the phenotype was typical of ACS, the entire coding sequence of KIF7 and the flanking intronic segments were amplified by using primers designed by Primer3 and subsequently sequenced bidirectionally using standard Sanger sequencing (primers and PCR conditions are available upon request). Sequence analysis revealed a KIF7 homozygous truncating mutation (NM_198525.2:c.2272G>T, p.E758*) (Figure 2). In Cases 2 and 3, candidate genes for sequencing were determined by using autozygosity mapping as described before.5 Briefly, the entire set of autozygous intervals per genome (autozygome) was determined for each index by using stretches of homozygosity (⩾2 Mb) as a surrogate after performing genomewide genotyping on the Axiom (Affymetrix, Santa Clara, CA, USA) single-nucleotide polymorphism platform and subsequent search for homozygous intervals by using AutoSNPa (dna.leeds.ac.uk/autosnpa). Genes within the autozygome were interrogated for candidacy based on the clinical features by using the Genomic Oligoarray and single-nucleotide polymorphism array evaluation tool v2.0.6 In Case 2, autozygome-guided candidate gene analysis linked this patient to KIF7, and gene sequencing analysis revealed a homozygous truncating mutation (NM_198525.2:c.3331C>T, p.R1111*). In Case 3, autozygome analysis revealed KIF7 as a candidate gene and sequencing confirmed the presence of a homozygous trunscating mutation (NM_198525.2:c.2896_2897 del, p.Ala966Profs*81) (Figure 2). Segregation with the disease was confirmed for all three mutations within the respective families.

KIF7 gene schematic representation with a snippet of the sequence chromatogram for each of the three mutations.
KIF7 encodes a 1343 amino acid protein, a member of a family of 14 kinesin proteins known to be ‘motor proteins’, molecular proteins that hydrolyze adenosine triphosphate, converting its chemical energy into mechanical energy to aid their transport along microtubules.7,8 KIF7 is a cilia-associated protein that plays an important role in the Sonic Hedgehog signaling pathway.9–11 Specifically, KIF7 functions as a negative regulator of the Sonic Hedgehog pathway by preventing an inappropriate activation of Gli2, and as a positive regulator by preventing the processing of Gli3 into its repressor form.12,13
KIF7 mutations have been implicated in the pathogenesis of ACS, a ciliopathy with strong central nervous system involvement in the form of intellectual disability with total or partial corpus callosum absence, in addition to craniofacial dysmorphism and polydactyl.4,14 Hydrolethalus can be considered a more severe form of ACS with a lethal brain phenotype encompassing hydrocephaly and anencephaly. Mutations in KIF7 have been identified in patients with this phenotype, in addition to HYLS1.4,15 Interestingly, KIF7 mutations have also been reported in Joubert syndrome albeit rarely.16
Of the 39 cases of ACS reported so far, 22 of them have atypical features with regard to either a wider clinical spectrum or a milder phenotype.17–25 However, the very mild presentation of Case 3 that is limited to intellectual disability and dysgenesis of corpus callosum appears to be unique and raises an interesting question of the contribution of KIF7 to non-syndromic intellectual disability with corpus callosum agenesis. To address this, we sequenced KIF7 in 11 cases with corpus callosum agenesis and intellectual disability but did not identify any mutation. Thus, while this case demonstrates that KIF7 mutation can present very mildly as intellectual disability with corpus callosal dysgenesis, its contribution to this phenotype appears to be rare.
In summary, we show that KIF7 mutations can result in clinical phenotypes that span a wide spectrum and we expand that spectrum further to include intellectual disability with corpus callosal dysgenesis.
References
Baker K, Beales PL . Making sense of cilia in disease: the human ciliopathies. Am J Med Genet C Semin Med Genet 2009; 4: 281–295.
Waters AM, Beales PL . Ciliopathies: an expanding disease spectrum. Pediatr Nephrol 2011; 26: 1039–1056.
Shaheen R, Faqeih E, Alshammari MJ, Swaid A, Al-Gazali L, Mardawi E et al. Genomic analysis of Meckel–Gruber syndrome in Arabs reveals marked genetic heterogeneity and novel candidate genes. Eur J Hum Genet 2012; 21: 762–768.
Putoux A, Thomas S, Coene KL, Davis EE, Alanay Y, Ogur G et al. KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat Genet 2011; 43: 601–606.
Alkuraya FS . Homozygosity mapping: one more tool in the clinical geneticist's toolbox. Genet Med 2010; 12: 236–239.
Wierenga KJ, Jiang Z, Yang AC, Mulvihill JJ, Tsinoremas NF . A clinical evaluation tool for SNP arrays, especially for autosomal recessive conditions in offspring of consanguineous parents. Genet Med 2012; 15: 354–360.
Klejnot M, Kozielski F . Structural insights into human Kif7, a kinesin involved in Hedgehog signalling. Acta Crystallogr D Biol Crystallogr 2012; 68 (Pt 2): 154–159.
Miki H, Setou M, Kaneshiro K, Hirokawa N . All kinesin superfamily protein, KIF, genes in mouse and human. Proc Natl Acad Sci USA 2001; 98: 7004–7011.
Varjosalo M, Taipale J . Hedgehog: functions and mechanisms. Genes Dev 2008; 22: 2454–2472.
Endoh-Yamagami S, Evangelista M, Wilson D, Wen X, Theunissen JW, Phamluong K et al. The mammalian Cos2 homolog Kif7 plays an essential role in modulating Hh signal transduction during development. Curr Biol 2009; 19: 1320–1326.
Bay SN, Caspary T . What are those cilia doing in the neural tube? Cilia 2012; 1: 19.
Liem KF Jr, He M, Ocbina PJ, Anderson KV . Mouse Kif7/Costal2 is a cilia-associated protein that regulates Sonic hedgehog signaling. Proc Natl Acad Sci USA 2009; 106: 13377–13382.
Liu CZ, Yang JT, Yoon JW, Villavicencio E, Pfendler K, Walterhouse D et al. Characterization of the promoter region and genomic organization of GLI, a member of the Sonic Hedgehog-Patched signaling pathway. Gene 1998; 209 (1-2): 1–11.
Schinzel A, Schmid W . Hallux duplication, postaxial polydactyly, absence of the corpus callosum, severe mental retardation, and additional anomalies in two unrelated patients: a new syndrome. Am J Med Genet 1980; 6: 241–249.
Mee L, Honkala H, Kopra O, Vesa J, Finnila S, Visapaa I et al. Hydrolethalus syndrome is caused by a missense mutation in a novel gene HYLS1. Hum Mol Genet 2005; 14: 1475–1488.
Dafinger C, Liebau MC, Elsayed SM, Hellenbroich Y, Boltshauser E, Korenke GC et al. Mutations in KIF7 link Joubert syndrome with Sonic Hedgehog signaling and microtubule dynamics. J Clin Invest 2011; 121: 2662–2667.
Ali BR, Silhavy JL, Akawi NA, Gleeson JG, Al-Gazali L . A mutation in KIF7 is responsible for the autosomal recessive syndrome of macrocephaly, multiple epiphyseal dysplasia and distinctive facial appearance. Orphanet J Rare Dis 2012; 7: 27.
Aykut A, Cogulu O, Ekmekci AY, Ozkinay F . An additional manifestation in acrocallosal syndrome: temporal lobe hypoplasia. Genet Couns 2008; 19: 237–240.
Fryns JP, Spaepen A, Grubben C, van den Berghe H, Casaer P . The variable clinical spectrum and mental prognosis of the acrocallosal syndrome. J Med Genet 1991; 28: 214–215.
Koenig R, Bach A, Woelki U, Grzeschik KH, Fuchs S . Spectrum of the acrocallosal syndrome. Am J Med Genet 2002; 108: 7–11.
Philip N, Apicella N, Lassman I, Ayme S, Mattei JF, Giraud F . The acrocallosal syndrome. Eur J Pediatr 1988; 147: 206–208.
Putoux A, Nampoothiri S, Laurent N, Cormier-Daire V, Beales PL, Schinzel A et al. Novel KIF7 mutations extend the phenotypic spectrum of acrocallosal syndrome. J Med Genet 2012; 49: 713–720.
Schinzel A . The acrocallosal syndrome in first cousins: widening of the spectrum of clinical features and further support for autosomal recessive inheritance. J Med Genet 1988; 25: 332–336.
Temtamy SA, Meguid NA . Hypogenitalism in the acrocallosal syndrome. Am J Med Genet 1989; 32: 301–305.
Turolla L, Clementi M, Tenconi R . How wide is the clinical spectrum of the acrocallosal syndrome? Report of a mild case. J Med Genet 1990; 27: 516–518.
Acknowledgements
We thank the families for their participation. We also thank the Genotyping and Sequencing Core Facilities at KFSHRC for their technical help. This work was funded in part by KACST Grant 13-BIO1113-20 (FSA).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Barakeh, D., Faqeih, E., Anazi, S. et al. The many faces of KIF7. Hum Genome Var 2, 15006 (2015). https://doi.org/10.1038/hgv.2015.6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2015.6
