
Abstract
Warburg micro syndrome is an autosomal recessive disease where patients present with optic, neurologic and genital symptoms. Until now, four disease genes for Warburg micro syndrome, RAB3GAP1, RAB3GAP2, RAB18 and TBC1D20, have been identified. Here, we report two novel homozygous RAB3GAP1 mutations (c.22G>T, p.Glu8* and c.1353delA, p.Pro452Hisfs*5) in two consanguineous families by whole-exome sequencing.
Similar content being viewed by others
Warburg micro syndrome (WARBM) is a rare, genetically heterogeneous, autosomal recessive syndrome. Patients with WARBM present with severe mental retardation, brain anomalies (polymicrogyria and corpus callosum hypoplasia), craniofacial features (microcephaly, hairy forehead, large anteverted ear, broad nasal root and micrognathia), ocular defects (congenital cataract, microphthalmia and microcornea), spasticity leading to contracture, congenital hypotonia and hypogonadism.1–4 The four WARBM subtypes (1, 2, 3 and 4) identified to date are caused by mutations in RAB3GAP1 (NM_001172435), RAB3GAP2 (NM_012414), RAB18 (NM_001256410) and TBC1D20 (NM_144628), respectively and are clinically indistinguishable.5–10 Here we analyze two unrelated WARBM patients to determine the underlying genetic abnormality.
Patient II-1 in family 1 (patient 1) is a 4-year-old Iranian girl born to consanguineous parents. She was born at 33 weeks of gestation after premature rupture of membranes without asphyxia (Apgar scores were 9 and 10 at 1 and 5 min, respectively). Her birth weight, length and head circumference were 1,850 g (7th centile), 49 cm (<5th centile) and 31 cm (7th centile), respectively. At birth, she showed characteristic craniofacial features (microcephaly, bitemporal narrowing, soft cleft palate, small mouth, micrognathia and large ears), ocular symptoms (bilateral cataract, microphthalmia and microcornea) and pectus carinatum. At 1 year of age, severe mental retardation, cerebral palsy, axial hypotonia and peripheral spasticity were recognized, with no seizures. At 17 months, a brain FLAIR-image revealed dysmyelination of the white matter implying severe hypoxic-ischemic encephalopathy, and the dysgenesis of corpus callosum. At 19 months, she showed mild bilateral conductive hearing loss detected by auditory brainstem response test. Currently at 4 years, she cannot sit independently. Neither parents have hearing impairments, pectus carinatum or soft cleft palates. Chromosome analysis of the patient was normal.
Patient II-3 in family 2 (patient 2) is an Indian girl, who is the third child of two healthy first cousins (Figure 1a). She had two unaffected siblings. She was born at 38 weeks of gestation by repeat cesarean section after an uneventful pregnancy. Her birth weight was 2,000 g (<5th centile). At 16 months of age, her weight was 6.2 kg (<5th centile), length 72 cm (<5th centile) and head circumference was 40 cm (<5th centile). She showed severe mental retardation, spoke only ‘mama’, ‘papa’ and ‘dada’ with hypotonia, exaggerated deep tendon reflexes in both legs, and hearing impairment. She had trigonocephaly, bilateral low-set prominent ears with anteriorly angulated, broad nasal root, micrognathia and a high-arched palate, and bilateral proximal placement of the thumbs and fifth toes. Microphthalmia, unresponsive pupils, microcornea and bilateral cataracts with very small vitreous cavities, leading to complete blindness were also noted. Hematological examination, thyroid, liver and renal function, serum calcium, ammonia, lactate, pyruvate and TORCH (toxoplasmosis, rubella, cytomegalovirus and herpes virus) were all normal at 16 months of age. Echocardiography, abdominal ultrasonography and skeletal survey were all normal, chromosomes were normal, and no hearing impairments were present in other family members.

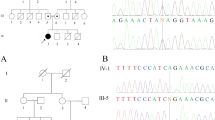
Familial pedigrees and mutations. (a) Family pedigrees with consanguinity. Black and white symbols are affected and unaffected, respectively. (b) Electropherograms of patients and their unaffected parents. The altered bases are shown in red characters.
The affected individuals from families 1 and 2 and their parents were analyzed. Peripheral blood samples were collected after obtaining written informed consent. DNA was extracted from peripheral blood leukocytes using the QIAamp DNA mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The Institutional Review Board of Yokohama City University School of Medicine approved this study.
Whole-exome sequencing was performed for the two affected individuals (II-1 in family 1, and II-3 in family 2) and for the unaffected parents of family 2 as previously reported.11,12 Briefly, genomic DNA (3 μg per sample) extracted from peripheral blood was sheared to 200 bp fragments using a Covaris S2 system (Covaris, Woburn, MA, USA). Genome partitioning was performed using a SureSelect Human All Exon Kit v5 (Agilent Technologies, Santa Clara, CA, USA). The prepared libraries were sequenced on a HiSeq2000 (Illumina, San Diego, CA, USA) with 101 bp paired-end reads with 7 bp index reads. Both reads were aligned to the human reference genome hg19 by Novoalign 3.00 (http://www.novocraft.com). The aligned reads were processed using Picard to remove polymerase chain reaction (PCR) duplicates (http://picard.sourceforge.net). The variants were called using the Genome Analysis Toolkit 2.4–4 (GATK; http://www.broadinstitute.org/gatk) with the GATK Best Practice Variant Detection v3 recommendations (http://www.broadinstitute.org/gatk/guide/topic?name=best-practices) and annotated using ANNOVAR (8 March 2012; http://www.openbioinformatics.org/annovar). Using these criteria, only variants located in the coding region and the adjacent 2 bp were identified. Common variants registered in dbSNP build 137 (minor allele frequency⩾0.01; http://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=316787363&g=snp137Common&hgTracksConfigPage=configure) were excluded. On the basis of the consanguinity in both families where autosomal recessive inheritance was considered, and homozygous variants were focused and validated by Sanger method (Supplementary Tables 1 and 2). PCR products were sequenced on an ABI3500xL sequencer (Applied Biosystems, Foster City, CA, USA) and analyzed using Sequencher 5.0 (Gene Codes Corporation, Ann Arbor, MI, USA).
The average-read depths for RefSeq coding DNA sequences were 88.5–99.1x in whole-exome sequencing. After in silico analysis, we focused on 16 and 13 homozygous candidate variants in patient 1 and patient 2, respectively (Supplementary Tables 3 and 4). Among these variants, homozygous truncating mutations were identified in RAB3GAP1 (NM_001143905): c.22G>T (p.Glu8*) in patient 1, and c.1353delA (p.Pro452Hisfs*5) in patient 2. Both mutations are novel. Each parents had a heterozygous mutation (Figure 1b). These mutations were absent in the NHLBI Exome Sequencing Project, the 1000 Genomes database and in our in-house exome database (n=575).
The RAB3GAP1 gene encodes RAB3 GTPase-activating protein, which has a role in converting the active GTP-bound form to the inactive GDP-bound form of RAB3 proteins, which in turn regulate hormone and neurotransmitter exocytosis.13,14 Among the four genes involved, RAB3GAP1 mutations are present in ~40% of WARBM patients, making it the most frequent cause. More than 50 mutations in RAB3GAP1 have been reported to date in the Human Genome Mutation Database (https://portal.biobase-international.com). Interestingly, they are mostly truncating mutations (nonsense, frameshift and splice-site) or microdeletions.5,6,9
Here we report two novel truncating RAB3GAP1 mutations (c.22G>T, p.Glu8* and c.1353delA, p.Pro452Hisfs*5) in two independent families. Both patients showed typical features of WARBM (Table 1). Of note, hearing impairment has been reported only in one patient,6 but was recognized in both patients described here, though we could not find any mutations that would cause hearing loss. Pectus carinatum and soft cleft palate, found in patient 1, have never been reported (Supplementary Table 5). As WARBM patients show variable skeletal abnormalities like pectus excavatum, kyphoscoliosis, hip dislocation and limb anomalies, pectus carinatum appears to be one of the skeletal phenotypes in WARBM.
In conclusion, we report two WARBM patients with novel RAB3GAP1 mutations. The hearing impairment, pectus carinatum and soft cleft palates seen in these patients have rarely or never been noted in WARBM.
References
References
Warburg M, Sjo O, Fledelius HC, Pedersen SA . Autosomal recessive microcephaly, microcornea, congenital cataract, mental retardation, optic atrophy, and hypogenitalism. Micro syndrome. Am J Dis Child 1993; 147: 1309–1312.
Graham JM Jr, Hennekam R, Dobyns WB, Roeder E, Busch D . MICRO syndrome: an entity distinct from COFS syndrome. Am J Med Genet A 2004; 128A: 235–245.
Abdel-Salam GM, Hassan NA, Kayed HF, Aligianis IA . Phenotypic variability in Micro syndrome: report of new cases. Genet Couns 2007; 18: 423–435.
Dursun F, Guven A, Morris-Rosendahl D . Warburg Micro syndrome. J Pediatr Endocrinol Metab 2012; 25: 379–382.
Aligianis IA, Johnson CA, Gissen P, Chen D, Hampshire D, Hoffmann K et al. Mutations of the catalytic subunit of RAB3GAP cause Warburg Micro syndrome. Nat Genet 2005; 37: 221–223.
Morris-Rosendahl DJ, Segel R, Born AP, Conrad C, Loeys B, Brooks SS et al. New RAB3GAP1 mutations in patients with Warburg Micro Syndrome from different ethnic backgrounds and a possible founder effect in the Danish. Eur J Hum Genet 2010; 18: 1100–1106.
Bem D, Yoshimura S, Nunes-Bastos R, Bond FC, Kurian MA, Rahman F et al. Loss-of-function mutations in RAB18 cause Warburg micro syndrome. Am J Hum Genet 2011; 88: 499–507.
Borck G, Wunram H, Steiert A, Volk AE, Korber F, Roters S et al. A homozygous RAB3GAP2 mutation causes Warburg Micro syndrome. Hum Genet 2011; 129: 45–50.
Handley MT, Morris-Rosendahl DJ, Brown S, Macdonald F, Hardy C, Bem D et al. Mutation spectrum in RAB3GAP1, RAB3GAP2, and RAB18 and genotype-phenotype correlations in warburg micro syndrome and Martsolf syndrome. Hum Mutat 2013; 34: 686–696.
Liegel RP, Handley MT, Ronchetti A, Brown S, Langemeyer L, Linford A et al. Loss-of-function mutations in TBC1D20 cause cataracts and male infertility in blind sterile mice and Warburg micro syndrome in humans. Am J Hum Genet 2013; 93: 1001–1014.
Saitsu H, Nishimura T, Muramatsu K, Kodera H, Kumada S, Sugai K et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat Genet 2013; 45: 445–449, 449e441.
Imagawa E, Osaka H, Yamashita A, Shiina M, Takahashi E, Sugie H et al. A hemizygous GYG2 mutation and Leigh syndrome: a possible link? Hum Genet 2014; 133: 225–234.
Fukui K, Sasaki T, Imazumi K, Matsuura Y, Nakanishi H, Takai Y . Isolation and characterization of a GTPase activating protein specific for the Rab3 subfamily of small G proteins. J Biol Chem 1997; 272: 4655–4658.
Fukuda M . Regulation of secretory vesicle traffic by Rab small GTPases. Cell Mol Life Sci 2008; 65: 2801–2813.
Data Citations
Miyake, Noriko, & Matsumoto, Naomichi HGV Database (2015) http://dx.doi.org/10.6084/m9.figshare.hgv.696
Miyake, Noriko, & Matsumoto, Naomichi HGV Database (2015) http://dx.doi.org/10.6084/m9.figshare.hgv.699
Acknowledgements
We thank the patients and their families for participating in this study. We also thank Ms. S Sugimoto and K Takabe for their technical assistance. This work was supported by grants from the Ministry of Health, Labour and Welfare (H Saitsu, N Miyake and N Matsumoto), a Grant-in-Aid for Scientific Research (A) (NM), a Grant-in-Aid for Scientific Research (B) (HS and NMi), a Grant-in-Aid for Scientific Research (C) (SM), a Grant-in-Aid for challenging Exploratory Research (HS) from the Japan Society for the Promotion of Science, the fund for Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems from the Japan Science and Technology Agency (NMa), a Grant-in-Aid for Scientific Research on Innovative Areas (Transcription Cycle) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (NMi and NMa), and the Takeda Science Foundation (Hs, NMi and NMa) and the Strategic Research Program for Brain Science (SRPBS) from Japan Agency for Medical Research and Development (AMED) (NMa).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information for this article can be found on the Human Genome Variation website (http://www.nature.com/hgv).
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Imagawa, E., Fukai, R., Behnam, M. et al. Two novel homozygous RAB3GAP1 mutations cause Warburg micro syndrome. Hum Genome Var 2, 15034 (2015). https://doi.org/10.1038/hgv.2015.34
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2015.34
