
Abstract
Epilepsy of infancy with migrating focal seizures (EIFMS) is a rare, early-onset epileptic encephalopathy characterized by polymorphous focal seizures. De novo mutations of KCNT1 have been identified in cases of this disorder. We encountered a sporadic patient with EIFMS, who suffered tonic convulsions at the age of 9 days. Using Sanger sequencing, we identified a de novo missense mutation of the same amino acid affected by a previously identified mutation, c.1420C>T (p.Arg474Cys).
Similar content being viewed by others

Epilepsy of infancy with migrating focal seizures (EIFMS), first described as migrating partial seizures of infancy (MMPSI) in 1995, is a rare, early-onset epileptic encephalopathy characterized by polymorphous focal seizures that commence within the first 6 months after birth.1 This clinical condition is classified as early infantile epileptic encephalopathy 14 (MIM #614959) by the online database Mendelian inheritance in man (OMIM; http://www.omim.org/). Seizures are pharmacoresistant and ictal electroencephalogram (EEG) discharges show migrating ictal foci. In 2012, mutations of the potassium channel, subfamily T, member 1 gene (KCNT1; MIM #608167) located at 9q34.3 were identified in familial patients with severe autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE; MIM #615005) associated with intellectual/psychiatric problems.2 Also, de novo mutations of KCNT1 were reported in 6 out of 12 unrelated patients (50%) with EIFMS,3 indicating the existence of clinical heterogeneity within KCNT1 mutations. Those results suggested that familial mutations and de novo mutations of KCNT1 correlate with ADNFLE and EIFMS, respectively. Different mutations of KCNT1 were identified by subsequent analyses in EIFMS patients.4 These findings suggested that KCNT1 is the major disease-associated gene for the EIFMS phenotype.
KCNT1 encodes a sodium-activated potassium (KNa) channel that is highly expressed in the nervous system.3 It is thought to regulate hyperpolarization following repetitive firing. The C-terminal cytoplasmic domain of KCNT1 interacts with a protein network, including the Fragile X mental retardation protein, thus stimulating the KCNT1 channel.5 Barcia et al.3 reported that mutations in the cytoplasmic C-terminal domain lead to constitutive activation of the KCNT1 channel. Our previous study reported two unrelated patients with EIFMS caused by a de novo missense mutation at the pore region of the KCNT1 channel.4
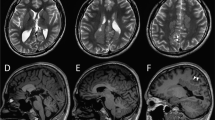
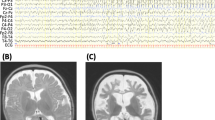
In the present study, we report on a 6-month-old Japanese male infant, the first child of nonconsanguineous healthy parents, born at 39 weeks of gestation using vacuum extraction because of diminished heart sounds. The child experienced an episode of apnea 4 days after delivery and tonic convulsions at day 9. Seizures evolved to frequent focal motor seizures that alternated from one side of the body to the other. Ictal EEGs showed asynchronous multifocal spikes derived from both brain hemispheres (Figure 1a). Seizures were intractable and easily led to status epilepticus, occurring in clusters. The child’s psychomotor development was markedly delayed, he was confined to his bed and exhibited poor responses to his surroundings. Brain magnetic resonance imaging (MRI) conducted at 6 months of age showed delayed myelination and thin corpus callosum (Figure 1b). These findings have been supported by a cohort study of 14 patients with EIFMS, one-third of the patients exhibited delayed myelination with white matter hyperintensity revealed by a brain MRI6 and 3 among the 6 patients with KCNT1 mutations showed a thin corpus collosum.3 On the basis of these clinical features, the child was diagnosed with EIFMS.

Patient clinical information. (a) Ictal electroencephalogram (EEG) examined at 6 months. A short EEG seizure discharge begins at the right frontal region. (b–d) Brain magnetic resonance imaging examined at 6 months. Extracerebral spaces can be seen in axial images, (b and c) possibly indicating brain atrophy. Both (b) T1- and (c) T2-weighted images show high intensity in the deep white matter, indicating delayed myelination. (d) The corpus callosum is thin in the T1-weighted sagittal image.
This study has been approved by Ethics Committee of Tokyo Women’s Medical University. Upon obtaining written informed consent from the patient’s family, mutation analysis using standard Sanger sequencing was performed for all exons of KCNT1 and a heterozygous missense mutation (c.1420C>T, p.Arg474Cys) identified in exon 15 was the only variant found (Figure 2). Because both parents did not exhibit this mutation, it was determined to be de novo in origin. This alteration has not been previously reported and is not found in the single-nucleotide variation database (https://genome.ucsc.edu/). The affected amino acid is highly conserved among species (Figure 2). A de novo missense mutation at the same amino-acid, p.Arg474His, has been reported in a patient with EIFMS.3 The clinical features of both patients with amino-acid changes at p.Arg474 are summarized in Table 1. Because both patients shared quite similar clinical characteristics, we concluded that p.Arg474Cys identified in this study could be pathogenic.

Electropherogram and corresponding genomic data. The electropherogram of the KCNT1 exon 15 indicates a heterozygous missense mutation, c.1420C>T (p.Arg474Cys), in KCNT1. The affected residue is conserved across species. Although there are some single-nucleotide variants (SNVs) proximal to the identified mutation, the same SNVs are not found in the database.
In conclusion, we identified a novel de novo missense mutation of KCNT1 in a patient with sporadic EIFMS. This finding provides additional evidence to better understand KCNT1 pathogenesis.
References
References
Coppola G, Plouin P, Chiron C, Robain O, Dulac O . Migrating partial seizures in infancy: a malignant disorder with developmental arrest. Epilepsia 1995; 36: 1017–1024.
Heron SE, Smith KR, Bahlo M, Nobili L, Kahana E, Licchetta L et al. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet 2012; 44: 1188–1190.
Barcia G, Fleming MR, Deligniere A, Gazula VR, Brown MR, Langouet M et al. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet 2012; 44: 1255–1259.
Ishii A, Shioda M, Okumura A, Kidokoro H, Sakauchi M, Shimada S et al. A recurrent KCNT1 mutation in two sporadic cases with malignant migrating partial seizures in infancy. Gene 2013; 531: 467–471.
Brown MR, Kronengold J, Gazula VR, Chen Y, Strumbos JG, Sigworth FJ et al. Fragile X mental retardation protein controls gating of the sodium-activated potassium channel slack. Nat Neurosci 2010; 13: 819–821.
McTague A, Appleton R, Avula S, Cross JH, King MD, Jacques TS et al. Migrating partial seizures of infancy: expansion of the electroclinical, radiological and pathological disease spectrum. Brain 2013; 136: 1578–1591.
Data Citations
Yamamoto, Toshiyuki HGV Database (2014) http://dx.doi.org/10.6084/m9.figshare.hgv.524
Acknowledgements
The authors express their gratitude to the patient and his parents for their cooperation. This work was supported by a Grant-in-Aid for Scientific Research from the Health Labor Sciences Research Grants program of the Ministry of Health, Labor, and Welfare, Japan (TY).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Shimada, S., Hirano, Y., Ito, S. et al. A novel KCNT1 mutation in a Japanese patient with epilepsy of infancy with migrating focal seizures. Hum Genome Var 1, 14027 (2014). https://doi.org/10.1038/hgv.2014.27
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2014.27
This article is cited by
-
Interaction Between HCN and Slack Channels Regulates mPFC Pyramidal Cell Excitability in Working Memory Circuits
Molecular Neurobiology (2023)
-
Sleep related hyper motor epilepsy (SHE): a unique syndrome with heterogeneous genetic etiologies
Sleep Science and Practice (2019)
