
Abstract
Epilepsy of Infancy with Migrating Focal Seizures (EIMFS) is a rare developmental and epileptic encephalopathy (DEEs) with unknown etiology, and poor prognosis. In order to explore new genetic etiology of EIMFS and new precision medicine treatment strategies, 36 children with EIMFS were enrolled in this study. 17/36 cases had causative variants across 11 genes, including 6 novel EIMFS genes: PCDH19, ALDH7A1, DOCK6, PRRT2, ALG1 and ATP7A. 13/36 patients had ineffective seizure control, 14/36 patients had severe retardation and 6/36 patients died. Of them, the genes for ineffective seizure control, severe retardation or death include KCNT1, SCN2A, SCN1A, ALG1, ATP7A and WWOX. 17 patients had abnormal MRI, of which 8 had ineffective seizure control, 7 had severe retardation and 4 died. 13 patients had hypsarrhythmia, of which 6 had ineffective seizure control, 6 had severe retardation and 2 died. Also, 7 patients had burst suppression, of which 1 had ineffective seizure control, 3 had severe retardation and 3 died. This study is the first to report that ALDH7A1, ATP7A, DOCK6, PRRT2, ALG1, and PCDH19 mutations cause the phenotypic spectrum of EIMFS to expand the genotypic spectrum. The genes KCNT1, SCN2A, SCN1A, ALG1, ATP7A and WWOX may be associated with poor prognosis. The patients presenting with MRI abnormalities, hypsarrhythmia and burst suppression in EEG may be associated with poor prognosis.
Similar content being viewed by others


Epilepsy of infancy with migrating focal seizures (EIMFS) syndrome, which was first reported in 19951, is a rare developmental and epileptic Encephalopathy (DEEs) with poor prognosis, presenting with focal seizures in the first year of life. The main clinical features are seizure onset in the first 6 months of life, occurrence of almost continuous migrating polymorphous focal seizures, combined with multifocal ictal electroencephalography (EEG) discharges, and progressive deterioration of psychomotor development2.
Previously, EIMFS was reported as an epilepsy syndrome with unknown etiology and poor prognosis3. Currently, in pace with the continuous development of genetic techniques, an increasing number of novel pathogenic genes have been shown to be related to EIMFS, including GABRA1, GABRB1, GABRB3, KCNQ2, BRAT1, HCN1, SCN8A, SMC1A, TBC1D24, QARS, ATP1A3, CDKL5, PIGA, ITPA, AIMP1, KARS, WWOX, KCNT1, TBC1D24, SLC25A22, SCN1A, SCN2A, SLC12A5, PLCB1 and PNPO2,4,5,6. Correspondingly, precision medicine treatments were adopted and partly improved patient outcomes. For example, GoF SCN2A variants present with EIMFS might respond to sodium channel blockers, while LoF SCN2A variants present West syndrome, myoclonic atonic epilepsy, and unclassified DEEs should avoid sodium channel blockers7. Early quinidine intervention associated with heart monitoring and control of blood levels is among the critical factors for therapy effectiveness in EIMFS patients with KCNT1 gain-of-function mutations8. Hence, this study included 36 patients who were diagnosed with EIMFS; the clinical features, etiology, treatment strategies and outcomes of these patients were determined to explore new genetic etiology and new precision medicine treatment strategies.
Results
Clinical phenotypic spectrum
A total of 36 patients were enrolled in this study, and all patients fulfilled the inclusion criteria. We presented the phenotypic summary of the cohort (Table 1). Among them, 25 (69.4%) were male, and 11 (30.6%) were female. Eleven patients (30.6%) had seizure onset at 4 hours to 3 days after birth. Ten (27.8%) patients had a family history of epilepsy, febrile seizure or mental retardation, or stillbirth. There were 10 (27.8%) patients with congenital heart disease, 8 (22.2%) patients with perinatal history, 1 (2.8%) patients with intracranial hemorrhage, 1 (2.8%) patients with inguinal hernia, 3 (8.3%) patient with intracranial infection, 1 (2.8%) patient with Menkes disease and 1 (2.8%) patient with congenital disorder of glycosylation, type Ik.
Seizure type at onset included migrating focal seizure (36/36, 100%); tonic seizure (21/36, 58.3%); spasms (8/36, 22.2%); clonic seizure (5/36, 13.9%) and tonic-clonic, autonomic seizures and others (8/36, 22.2%). Hypsarrhythmia (classic or modified) was seen in 13 (13/36, 36.1%) patients, and a burst-suppression pattern was observed in 7 (7/36, 19.4%) patients. All patients underwent brain MRI and/or CT examinations, 17 of whom had abnormal brain imaging findings. Diffuse cerebral atrophy (with an increase in extra axial fluid) was observed in 3 children. Four cases demonstrated forehead dysplasia or widened brain space, 3 presented with hippocampal sclerosis, 3 presented with encephalomalacia foci, 1 presented with dysplasia of the corpus callosum, 5 presented with subependymal cyst, 1 presented with pachygyria and 1 presented with meningeal reinforcement. Specifically, one of the 3 children with cerebral atrophy was accompanied with hippocampal sclerosis. One child had encephalomalacia foci, hippocampal sclerosis, subependymal cyst and widened brain space.
The etiological composition of the 36 patients in this cohort was determined by the International League Against Epilepsy (ILAE) 2017 etiological classification criteria. Accordingly, there were 17 (17/36, 47.2%) patients with genetic etiology, of which 6 (6/17, 35.3%) had severe retardation, 3 (3/17, 17.6%) died and 2 (2/17, 11.8%) was normal. There were 2 (2/36, 5.6%) patients with structural etiology, both of them had severe retardation. Three patient (3/36, 8.3%) had infectious etiology, and 1 died. There were two (2/36, 5.6%) patients with metabolic etiology (both cases were inherited), of which 1 had severe retardation and 1 died. There was no immune-mediated etiology and 14 (14/36, 38.9%) cases for unknown etiology, of which 7 (7/14, 50.0%) had severe retardation and 2 (2/14, 14.3%) were normal.
Genotypic spectrum
There were 26 (26/36, 72.2%) patients who underwent genetic testing, including 15 (15/26, 57.7%) with whole exome sequencing (WES), 11 (11/26, 42.3%) with an epilepsy gene panel, 3 (3/26, 11.5%) with mitochondrial genome sequencing, 1 (1/26, 3.8%) with chromosome karyotype analysis and 1 (1/26, 3.8%) with copy number variant (CNV) analysis. Specifically, one of the 2 patients with mitochondrial genome sequencing and WES, one of the 1 patient with mitochondrial genome sequencing, WES, chromosome karyotype analysis and CNV analysis. Seventeen (17/36, 47.2%) cases had causative variants across 11 genes (Table 2), including 6 novel EIMFS genes: de novol PCDH19; paternal ALDH7A1, DOCK6, PRRT2 and ALG1; maternal ALDH7A1, ATP7A, DOCK6 and ALG1. Five genes have been reported in previous literature, including de novo KCNT1, SCN2A, SCN1A, paternal PNPO, WWOX, maternal PNPO and WWOX. The most frequently implicated genes were KCNT1 (3/36, 8.3%) and SCN2A (6/36, 16.7%).
EIMFS phenotype-genotype correlation
Phenotypic data are summarized according to each gene in Table 3. We highlight the phenotypes of the five most frequently implicated genes for EIMFS here.
KCNT1 EIMFS patients (n = 3)
Three patients (3 males) with KCNT1 variants aged 1 year and 4 months to 2 years were studied (median 1 year 4months). The median seizure onset was 1 months (range 15 days to 4 months). All had focal seizures, 1 had tonic seizures, 2 had spasms seizures, and 2 had autonomic seizures. All had normal brain imaging findings. EEG showed seizure migration in all patients, hypsarrhythmia in 2/3, and burst suppression in 1/3. Two patients had severe developmental retardation, and one died. The age of death was 2 years.
SCN2A EIMFS (n=6)
There were 6 patients with SCN2A EIMFS studied at 1 month to 6 years of age (median 1 years 5 months) with median seizure onset at 2 days (range 1 day to 9 months). All had focal seizures, 4 had tonic seizures,2 had spasms seizures, and 1 had an autonomic seizure. A total of 2/6 had normal brain imaging findings, and 4/6 had abnormal imaging findings. EEG data showed seizure migration in all patients, hypsarrhythmia in 3/6, and burst suppression in 2/6. The effective drugs for 6 patients with seizure control were oxcarbazepine, carbamazepine, vigabatrin and levetiracetam. Three patients had severe developmental retardation, three patients had mild-moderate development retardation, and no patients died.
PNPO and ALDH7A1 EIMFS (n=2)
One patient (female) with PNPO variants was studied at 4 years and 4 months of age. Seizure onset was 2 days after birth. She had focal seizures and abnormal brain imaging findings. One patient (male) with ALDH7A1 variants was studied at 3 years and 10 months of age. Seizure onset was 1 month. He had focal and clonic seizures and normal brain imaging. There was no hypsarrhythmia or burst suppression in the EEG of these two patients. Vitamin B6 was effective in both patients. One patient had only mild development retardation, and one patient had normal development.
PRRT2 EIMFS (n=1)
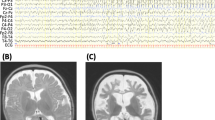
One patient (female) with PRRT2 variants was studied at 10.5 months of age. Seizure onset was 3 months. She had migarating focal seizures and normal brain imaging findings. Migarating focal seizures and no hypsarrhythmia or burst suppression was evident in the EEG data of the patient (Figure 1). Oxcarbazepine was effective in controlling seizures in this patient, who had normal development.

Migrating focal seizures was evident in the EEG data of the patient with PRRT2 gene mutation. (A) Interictal period showed multifocal discharge; (B1) Focal seizures originating in the right occipital and posterior temporal regions; (B2) More than 10 s later, the epileptiform electrical seizure in the right posterior occipital temporal region relieved, and electrical rhythm changes appeared in the right central region. (B3) Convulsions of the left upper limb, simultaneous electrical seizures in the right central region; (C1) Focal seizures originating in the right occipital and posterior temporal regions; (C2) Focal seizures migrate to the left central region.
Treatment
Anti seizure medication effects on seizures were analyzed in 36 patients with EIMFS (Table 3). In addition to classical anti seizure medication, vitamin B6 and adrenocorticotropic hormones (ACTH) were used to treat EIMFS patients. Vitamin B6 allowed patients with ALDH7A1 and PNPO mutations to achieve seizure-free status, oxcarbazepine was effective for patients with SCN2A, ATP7A, WWOX, and PRRT2 mutations, and ACTH was partly effective for DOCK6 mutation patients with spasms and hypsarrhythmia.
Outcome
We obtained data regarding seizure control and motor development for 36 patients: 14/36 (38.9%) patients were seizure free, and 9/36 (25.0%) patients had seizures reduced > 50%. However, 13/36 (36.1%) patients had ineffective seizure control. 14/36 (38.9%) patients had severe retardation, 13/36 (36.1%) patients had mild-moderate retardation, and 3/36 (8.3%) patients had normal development (Table 3). Our cohort had a high mortality rate: 6/36 (16.7%) patients died at a median age of 8 months (range 3 months to 2 years). In 3/6 patients who died, pathogenic variants were identified in the following genes: KCNT1 (1), SCN1A (1) and ALG1 (1), with 3 cases remaining unsolved. The genes for ineffective seizure control and severe retardation include KCNT1 (2), SCN2A (1), ATP7A (1) and WWOX (1). Genes associated with seizure-free, mild-moderate retardation or normal of mental and motor development included PRRT2 (1), SCN2A (3), ALDH7A1 (1) and PNPO (1).
Of the 36 patients with EIMFS, 19 had normal MRI findings, of which 9 (9/19, 47.4%) were seizure free, 9 (9/19, 47.4%) had ineffective seizure control, 7 (7/19, 36.8%) had severe retardation and 2 (2/19, 10.5%) died. Also, 17 had abnormal MRI, of which 5 (5/17, 29.4%) were seizure free, 8 (8/17, 47.1%) had ineffective seizure control, 7 (7/17, 41.2%) had severe retardation and 4 (4/17, 23.5%) died. The details are shown in Table 4.
Of the 36 patients with EIMFS, 13 had hypsarrhythmia, of which 5 (5/13, 38.5%) were seizure free, 6 (6/13, 46.1%) had ineffective seizure control, 6 (6/13, 46.1%) had severe retardation, 2 (2/13, 15.4%) died and 0 (0/13, 0%) were normal; also, 7 had burst suppression, of which 1 (1/7, 14.3%) were seizure free, 1 (1/7, 14.3%) had ineffective seizure control, 3 (3/7, 42.9%) had severe retardation, 3 (3/7, 42.9%) died and 0 (0/3, 0%) were normal. 3 patients had hypsarrhythmia and burst suppression, of which 1 (1/3, 33.3%) were seizure free, 1 (1/3, 33.3%) had ineffective seizure control, 2 (2/3, 66.7%) had severe retardation, 0 (0/3, 0%) died and 0 (0/3, 0%) were normal, and 19 had no hypsarrhythmia or burst suppression, of which 8 (8/19, 42.1%) were seizure free, 7 (7/19, 36.8%) had ineffective seizure control, 5 (5/19, 26.3%) had severe retardation, 2 (2/19, 10.5%) died and 3 (3/19, 15.8%) were normal. The details are shown in Table 5.
Discussion
EIMFS is characterized by nearly continuous seizures involving multiple independent areas of both hemispheres with arrested psychomotor development. It is an age-dependent, often overlooked syndrome among the epileptic encephalopathies that can occur within the first 6 months of life9. EIMFS has clinical phenotypic overlap with other epileptic encephalopathies in early infancy, such as early infantile epileptic encephalopathy, early myoclonic encephalopathy, and infantile spasms7,10. EIMFS has no specific neuroimaging changes reported in the current literature. We described the phenotype spectrum of 36 patients with EIMFS in this study. All patients had clinical seizure migration associated with a significant impact on development. We identified several key phenotypic features that have only been rarely reported in the EIMFS phenotypic spectrum: 11 (11/36, 30.6%) patients had suspected in utero seizures with postnatal seizures described between 4 hours and 3 days, 8 (8/36, 22.2%) patients had epileptic spasms, 7 (7/36, 19.4%) had a burst-suppression pattern in EEG activity, 1 patient had congenital disorder of glycosylation—type Ik, 1 patient had Menkes disease. Furthermore, it is mostly believed that the etiology of EIMFS is caused by genetic mutations9. According to our etiological analysis, the genetic etiology accounted for 17/36 cases, and structural etiology accounted for 2/36 cases. Analysis of etiology and prognosis results suggests that structural causes may be related to poor prognosis. Our data expand the clinical phenotype of EIMFS.
We describe the genotypic spectrum of 17 patients with EIMFS. We highlight the extensive genetic heterogeneity of EIMFS, which is similar to that in other epilepsy syndromes, such as infantile spasms (West syndrome) and Lennox-Gastaut syndrome, but with a considerably higher yield on current testing. We identified the etiology in 47.2% of our cohort, including 6 novel EIMFS genes. The most commonly involved genes were KCNT1 (8.3%) and SCN2A (16.7%), together explaining 25.0% of EIMFS cases. These studies highlight the importance of classifying a patient's epilepsy syndrome, which influences genetic testing and interpretation11. Epileptic spasms have only been rarely reported in EIMFS patients2. In this series, 8/36 patients (22.2%) had epileptic spasms, including those with KCNT1, SCN2A, SCN1A, DOCK6 and PCDH19 variants.
We discovered 6 novel EIMFS genes in our cohort - de novo PCDH19; paternal ALDH7A1, DOCK6, PRRT2 and ALG1; and maternal ALDH7A1, ATP7A, DOCK6 and ALG1 - encoding a wide range of proteins. These genes have not been described in patients with EIMFS, but they have been associated with other neurological diseases. Considering that one of our patients was a female and had a de novo mutation of the PCDH19 gene, which was consistent with the X-linked genetic classification and the common clinical phenotype of spasms seizure and delayed motor development12, the software predicted the likely pathogenicity of the mutation; thus, these findings suggested that the PCDH19 gene could be associated with EIMFS. ALDH7A1 gene is associated with autosomal recessive pyridoxine-dependent epilepsy13. Our patient's clinical phenotype was recurrent epileptic seizures, accompanied by developmental delay, and vitamin B6 treatment was effective. The inheritance pattern was consistent with family co-segregation, and the software predicted it as likely pathogenic. Therefore, it can be considered as the causative gene of EIMFS. In future studies, it is hoped that more case reports, or functional studies, can confirm the pathogenicity of this variant. DOCK6 gene is associated with autosomal recessive Adams-Oliver syndrome-2, which is characterized by aplasia cutis congenita (ACC) as well as terminal transverse limb defects (TTLD) in addition to variable involvement of the brain, eyes, and cardiovascular system14. Our patient had developmental delays and recurrent seizures, which may be related to brain development disorders. Therefore, we could not determine whether the DOCK6 gene was the causative gene of EIMFS, and further data are needed for verification. It has been previously thought that PRRT2-associated paroxysmal movement disorders (PRRT2-PxMD) are mostly manifested as infantile-onset epilepsy and post-adolescent movement disorders, and most of them have a good prognosis and do not affect cognitive development. In addition, PRRT2 pathogenic variants have been identified in other childhood-onset movement disorders and different types of seizures, suggesting that the understanding of the spectrum of PRRT2-PxMD is still evolving15. The patient with this mutation in our cohort was very interesting. He presented with very frequent cluster seizures when he was 3 months old, video-EEG demonstrated migrating focal seizures and typical background characteristics of EIMFS, and he experienced obvious cognitive retardation after the onset of epilepsy. He achieved seizure-free status after the administration of oxcarbazepine, and follow-up showed normal cognitive development. The patient had a paternal mutation of the PRRT2 gene—his father had symptoms of dystonia in his youth—and the software predicted the likely pathogenicity of the mutation. We considered that the PRRT2 gene was the causative gene of EIMFS, suggesting that PRRT-2 spectrum diseases could also manifest as a very severe epileptic encephalopathy phenotype, but after effective treatment, the disease process could be quickly reversed and the prognosis was good. In the 10th patient, two genetic variants were detected simultaneously, namely, WWOX and ATP7A. The clinical phenotypes of WWOX gene reported in the literature were autosomal recessive epileptic encephalopathy, early infantile 28, esophageal squamous cell carcinoma, somatic and spinocerebellar ataxia, and autosomal recessive 1216,17. The WWOX gene was EIMFS-related genes reported in previous literature, which theoretically can cause epileptic encephalopathy. The clinical phenotypes of ATP7A gene reported in the literature were X-linked Menkes disease, occipital horn syndrome, distal spinal muscular atrophy 318,19,20. This patient also had a significant decrease in blue copper protein, had hair and skin color changes and could have been diagnosed with Menkes disease. At present, the epileptic seizure forms of Menke's disease reported in the literature include focal clonic seizures, myoclonic, and tonic seizures, infantile spasms, and no migrating focal seizures have been reported21. We could not confirm whether it was a copathogenic gene. ALG1 gene is associated with autosomal recessive congenital disorder of glycosylation type Ik22,23, which manifested as feeding problems and diarrhea, muscular hypertonia, refractory seizures, recurrent episodes of apnea, cardiac and hepatic involvement and coagulation anomalies24. The clinical characteristics of this patient were consistent with the above core symptoms, the genetic results were consistent with the law of autosomal recessive inheritance, and the software predicted the likely pathogenicity of the mutation. We considered the ALG1 gene to be a newly pathogenic gene of EIMFS.
Most patients with EIMFS are refractory to anti-seizure medication, but some have shown good progression or a near satisfactory response to treatment. The anti-seizure medication used alone or in combination with one another that may achieve seizure control or reduction are potassium bromide, levetiracetam, ACTH, stiripentol, clonazepam, and rufinamide25. Mikati et al. reported that quinidine is effective in the treatment of patients with KCNT1 gene mutations in EIMFS26. Some studies have found that a correlation between age at disease onset, response to sodium channel blockers and the functional properties of mutations in children with SCN2A-related epilepsy. The use of sodium channel blockers was often associated with clinically relevant seizure reduction or seizure freedom in children with early infantile epilepsies (<3 months), whereas other antiepileptic drugs were less effective. In contrast, sodium channel blockers were rarely effective in epilepsies with later onset (≥3 months) and sometimes induced seizure worsening27. At present, there are few literature reports on the use of corresponding effective drug treatments for patients with different EIMFS gene mutations. In our cohort, we determined that vitamin B6 could allow patients with ALDH7A1 and PNPO mutations to achieve seizure-free status. Oxcarbazepine was effective for patients with SCN2A, ATP7A+WWOX, and PRRT2 mutations. Among our 6 children with SCN2A gene mutations, 4 of them were onset before 3 months of age, and the seizure control effect was good, which was consistent with the data reported in the literature. ACTH was partly effective for patients with DOCK6 mutations who had spasms and hypsarrhythmia.
While seizure outcomes and developmental prognoses are generally poor in EIMFS, there are rare reports of mildly affected patients28. In our study cohort, the incidence of poor prognosis was also relatively high; 6/36 (16.7%) patients died, and the related pathogenic genes were KCNT1, SCN1A, ALG1. 14/36 (38.9%) patients had severe retardation, and the genes for ineffective seizure control and severe retardation included KCNT1, SCN2A, WWOX and ATP7A. The results indicated that the related pathogenic genes KCNT1, SCN1A, ALG1, SCN2A, WWOX and ATP7A may be associated with ineffective seizure control and poor prognoses. While all patients experienced refractory epilepsy early in the course of the disease, 3/36 (8.3%) patients had normal mental and motor development. Genes associated with seizure-free, mild-moderate retardation or normal of mental and motor development included PRRT2, SCN2A, ALDH7A1, PCDH19 and PNPO.
In addition, we compared the association of MRI abnormalities, hypsarrhythmia and burst suppression in EEG with poor prognosis. The results found that patients with EIMFS characterized by abnormal MRI, hypsarrhythmia and burst suppression in EEG have a higher incidence of ineffective seizure control, severe retardation and a higher mortality rate. The results suggest that EIMFS patients who present with abnormal MRI, hypsarrhythmia and burst suppression in EEG may be associated with ineffective seizure control and poor prognosis. We need to further expand the sample to analyze and confirm these correlations.
Conclusions
In conclusion, our study expands the EIMFS clinical phenotype and genotype spectra. EIMFS patients who present with abnormal MRI findings, hypsarrhythmia and burst suppression in EEG may be associated with ineffective seizure control and poor prognosis. Etiological analysis showed that in addition to genetic mutations, structural etiology may be associated with poor prognosis. This study is the first to report that ALDH7A1, ATP7A, DOCK6, PRRT2, ALG1, and PCDH19 mutations cause the phenotypic spectrum of EIMFS. The genes KCNT1, SCN1A, ALG1, SCN2A, WWOX and ATP7A may be associated with ineffective seizure control and poor prognosis. Through early diagnosis with genetic tests and the administration of the corresponding precise treatment, the outcomes of EIMFS can be notably improved.
Methods
Participants and phenotyping
Our EIMFS cohort comprised 36 patients from Hunan Children's Hospital and Qilu Hospital of Shandong University. A multicenter retrospective case study was performed over a 10-year period (January 2010 to January 2020). The parents of the patients provided written, informed consent. This study was approved by the Medical Ethics Committee of Hunan Children’s Hospital and Qilu Hospital of Shangdong University.
According to the clinical characteristics of EIMFS described by Coppola1, the inclusion criteria are as follows: (1) onset within 6 months after birth; (2) migrating focal seizures at onset; (3) multifocal seizures intractable to conventional antiepileptic drugs; (4) the EEG during the onset period is characterized by multifocal discharges, migrating in one hemisphere or between the two hemispheres, involving multiple parts, and the clinical onset is closely related to the time and location of the EEG discharge; and (5) delayed developmental progress or signs of psychomotor regression associated with seizure onset. The phenotypic information of all patients was collated on clinical presentation, disease course, EEG, neuroimaging, treatment strategies and the results of neurometabolic and diagnostic genetic investigations. All patients were followed up every 1-6 months by telephone or outpatient department.
It should be noted in particular that one of our patients met the inclusion criteria except for the onset age of 9 months. Considering that EIMFS with onset age of 9 months has been reported in literatures, we still included this patient in our study cohort.
Genetic tests
Genetic testing was carried out using chromosome karyotype analysis, CNV analysis, mitochondrial genome sequencing, epilepsy gene panels and WES. CNV analysis was performed with the Illumina HumanOmniZhonghua-8 Bead Chip; Mitochondrial genome sequencing was subsequently performed on an Illumina HiSeq 2000 platform (Illumina, San Diego, CA, USA); details were provided by our team previously29. The epilepsy gene panel contained 265 epilepsy-associated genes was performed on methods previously reported by Lemke et al30. WES was also performed based on methods previously reported by our team29 by using Illumina HiSeq X Ten (Illumina, San Diego, CA, USA) with 150-bp paired-end reads.
Ethics approval and consent to participate
The study was performed in accordance with the Declaration of Helsinki, with the approval of the study protocol by an independent ethics committee or institutional review board at each site. All patients provided written informed consent before participation.
Consent for publication
All authors have approved the final article and its publication.
Data availability
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
Abbreviations
- ACTH:
-
Adrenocorticotropic hormones
- ACC:
-
Aplasia cutis congenita
- BFIE:
-
Benign familial infantile epilepsy
- CNV:
-
Copy number variant
- EEG:
-
Electroencephalography
- EIEE-9:
-
Early infantile epileptic encephalopathy-9
- HM:
-
Hemiplegic migraine
- EIMFS:
-
Epilepsy of Infancy with Migrating Focal Seizures
- HIE:
-
Hypoxic-ischemic encephalopathy
- ILAE:
-
International League Against Epilepsy
- IC:
-
Infantile convulsions
- MT I:
-
Mannosyltransferase I
- PRRT2-PxMD:
-
PRRT2-associated paroxysmal movement disorders
- PKD:
-
Paroxysmal kinesigenic dyskinesia
- TTLD:
-
Terminal transverse limb defects
- VUS:
-
Variant of unknown significance
- WES:
-
Whole exome sequencing
References
Coppola, G., Plouin, P., Chiron, C., Robain, O. & Dulac, O. Migrating partial seizures in infancy: A malignant disorder with developmental arrest. Epilepsia 36(10), 1017–1024 (1995).
McTague, A. et al. Migrating partial seizures of infancy: Expansion of the electroclinical, radiological and pathological disease spectrum. Brain 136(5), 1578–1591 (2013).
Coppola, G. Malignant migrating partial seizures in infancy: An epilepsy syndrome of unknown etiology. Epilepsia 50, 49–51 (2009).
Ohba, C. et al. Early onset epileptic encephalopathy caused by de novo SCN8A mutations. Epilepsia 55(7), 994–1000 (2014).
De Filippo, M. R. et al. Lack of pathogenic mutations in six patients with MMPSI. Epilepsy Res. 108(2), 340–344 (2014).
Burgess, R. et al. The genetic landscape of epilepsy of infancy with migrating focal seizures. Ann. Neurol. 86(6), 821–831 (2019).
Bayat, A., Bayat, M., Rubboli, G., & Moller, R. S. Epilepsy syndromes in the first year of life and usefulness of genetic testing for precision therapy. Genes (Basel) 12(7) (2021).
Dilena, R. et al. Early treatment with quinidine in 2 patients with epilepsy of infancy with migrating focal seizures (EIMFS) due to gain-of-function KCNT1 mutations: Functional studies, clinical responses, and critical issues for personalized therapy. Neurotherapeutics 15(4), 1112–1126 (2018).
Rizzo, F. et al. Characterization of two de novo KCNT1 mutations in children with malignant migrating partial seizures in infancy. Mol. Cell Neurosci. 72, 54–63 (2016).
Coppola, G. Malignant migrating partial seizures in infancy. Handb. Clin. Neurol. 111, 605–609 (2013).
McTague, A., Howell, K. B., Cross, J. H., Kurian, M. A. & Scheffer, I. E. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 15, 304–316 (2016).
Dibbens, L. M. et al. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat. Genet. 40(6), 776–781 (2008).
Mills, P. B. et al. Mutations in antiquitin in individuals with pyridoxine-dependent seizures. Nat. Med. 12(3), 307–309 (2006).
Shaheen, R. et al. Recessive mutations in DOCK6, encoding the guanidine nucleotide exchange factor DOCK6, lead to abnormal actin cytoskeleton organization and Adams-Oliver syndrome. Am. J. Hum. Genet. 89(2), 328–333 (2011).
Ebrahimi-Fakhari D, Moufawad El AchkarC, Klein C. PRRT2-associated paroxysmal movement disorders 1993.
Mallaret, M. et al. The tumour suppressor gene WWOX is mutated in autosomal recessive cerebellar ataxia with epilepsy and mental retardation. Brain 137(Pt 2), 411–419 (2014).
Abdel-Salam, G. et al. The supposed tumor suppressor gene WWOX is mutated in an early lethal microcephaly syndrome with epilepsy, growth retardation and retinal degeneration. Orphanet. J. Rare Dis. 9, 12 (2014).
Vulpe, C., Levinson, B., Whitney, S., Packman, S. & Gitschier, J. Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase. Nat. Genet. 3(1), 7–13 (1993).
Kaler, S. G. et al. Occipital horn syndrome and a mild Menkes phenotype associated with splice site mutations at the MNK locus. Nat. Genet. 8(2), 195–202 (1994).
Kennerson, M. L. et al. Missense mutations in the copper transporter gene ATP7A cause X-linked distal hereditary motor neuropathy. Am. J. Hum. Genet. 86(3), 343–352 (2010).
Verrotti, A., Carelli, A. & Coppola, G. Epilepsy in children with Menkes disease: A systematic review of literature. J. Child Neurol. 29(12), 1757–1764 (2014).
Schwarz, M. et al. Deficiency of GDP-Man:GlcNAc2-PP-dolichol mannosyltransferase causes congenital disorder of glycosylation type Ik. Am. J. Hum. Genet. 74(3), 472–481 (2004).
Harshman, L. A. et al. Congenital nephrotic syndrome in an infant with ALG1-congenital disorder of glycosylation. Pediatr Int. 58(8), 785–788 (2016).
Rohlfing, A. K. et al. ALG1-CDG: A new case with early fatal outcome. Gene 534(2), 345–351 (2014).
Caraballo, R., Pasteris, M. C., Fortini, P. S. & Portuondo, E. Epilepsy of infancy with migrating focal seizures: Six patients treated with bromide. Seizure 23(10), 899–902 (2014).
Mikati, M. A. et al. Quinidine in the treatment of KCNT1-positive epilepsies. Ann. Neurol. 78(6), 995–999 (2015).
Wolff, M. et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 140(5), 1316–1336 (2017).
Marsh, E., Melamed, S. E., Barron, T. & Clancy, R. R. Migrating partial seizures in infancy: Expanding the phenotype of a rare seizure syndrome. Epilepsia 46, 568–572 (2005).
Yang, H. et al. The study of genetic susceptibility and mitochondrial dysfunction in mesial temporal lobe epilepsy. Mol. Neurobiol. 57(9), 3920–3930 (2020).
Lemke, J. R. et al. Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia 53(8), 1387–1398 (2012).
Acknowledgements
We wish to thank the patients for the participation in this study.
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 82171453, 81671297), Natural Science Foundation of Hunan Province (2021JJ30393), Huxiang Youth Talent Support Program (2021RC3117) and Key projects supported by Hunan Provincial Health Commission (202203074966).
Author information
Authors and Affiliations
Contributions
H.Y. and X.Y. conducted the literature review and drafted the manuscript. F.C., S.G. and S.Y. made substantial contributions to the conception and interpretation of data. L.W. was responsible for revising the manuscript critically and gave final approval of the version to be published.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, H., Yang, X., Cai, F. et al. Analysis of clinical phenotypic and genotypic spectra in 36 children patients with Epilepsy of Infancy with Migrating Focal Seizures. Sci Rep 12, 10187 (2022). https://doi.org/10.1038/s41598-022-13974-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-13974-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
