
Abstract
Allan–Herndon–Dudley syndrome (AHDS) is a neurodevelopmental disorder that manifests as intellectual disability and motor developmental delay. Thyroid hormone transporter dysfunction due to SLC16A2 mutation is the underlying cause of this disorder. We identified a novel (P537del) and a recurrent (A150V) SLC16A2 mutation in Japanese AHDS patients from two different families. A150V co-segregated with S33P. Both patients showed similar clinical features including severe neurological features and delayed myelination. Thyroid function showed a common finding of elevated T3 levels. No clear genotype–phenotype correlation was observed in patients with SLC16A2 alterations.
Similar content being viewed by others

Allan–Herndon–Dudley syndrome (AHDS; MIM #300523) is recognized as a neurodevelopmental disorder that manifests in intellectual disability and motor developmental delay and is associated with infantile hypotonia. In 2004, the solute carrier family 16 member 2 gene (SLC16A2) was identified as the gene responsible for this disorder.1 SLC16A2 encodes a T3- and T4-transporter that is expressed in neuronal cells (monocarboxylate transporter 8; MCT8). MCT8 plays a decisive role in the transport of T3 into neurons.2 Therefore, the abnormal thyroid function observed in AHDS patients is the consequence of transporter dysfunction in neuronal cells.
In 2009, SLC16A2 mutations were identified in a number of patients initially presenting with mimicking phenotypes of Pelizaeus–Merzbacher disease (PMD) during the infantile period, in association with delayed myelination.3,4 Similar to PMD, AHDS is inherited as an X-linked recessive trait because SLC16A2 is located on Xq13.2. Many SLC16A2 mutations have been reported.5 In the present study, we identified SLC16A2 mutations in two different patients who initially presented with phenotypes mimicking those of PMD.
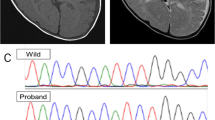
Patient 1 is a 7-month-old Japanese boy, who was born at 41 weeks of gestation with a birth weight of 3,040 g (mean). He was the only child of non-consanguineous parents. The family had no history of neuromuscular disorders. The pregnancy was not remarkable, and no asphyxic events occurred during birth. Although he showed social smiling, he did not show age-appropriate head control at the 5-month health checkup and was referred to our hospital. He showed hypertonus of his extremities and often displayed an asymmetrical tonic neck reflex posture with opisthotonus. He could neither turn over nor sit. Laboratory examinations revealed abnormal thyroid function with a normal TSH level of 3.647 μg/ml (normal range: 0.46–3.73), a high free T3 level of 7.73 pg/ml (normal range: 2.51–4.12) and a low free T4 level of 0.52 ng/dl (normal range: 0.88–1.5). Brain magnetic resonance imaging (MRI) of the patient at the age of 7 months showed markedly delayed myelination and mildly reduced brain volume (Figure 1a, b).

T1- and T2-weighted axial images of patient 1 examined at 7 months of age (a and b, respectively). Dilatation of the extra-cerebrum space and lateral ventricles is indicative of reduced brain volume. The genu of the corpus callosum is thin. Myelination is noted only in the posterior limb of the internal capsule and in the optic radiations, as shown in both of the T1- and T2-weighted images. T1- and T2-weighted axial images of patient 2 examined at 17 months of age (c and d, respectively). The extra-cerebrum space is remarkable. The T1-weighted image shows a normal myelination pattern with high intensity in the white matter (c); however, the T2-weighted image shows a delayed myelination pattern with low intensity only in the genu of the corpus callosum, anterior and posterior limbs of the internal capsule, and optic radiations (d).
Patient 2 is a 21-month-old Japanese boy who was born as the first child of healthy and non-consanguineous parents at 42 weeks of gestation with a birth weight of 3,686 g (90–97th percentile). At the age of 8 months, he was referred to our hospital because of a motor developmental delay. At that time, he had no head control and could not turn over. Physical examinations showed no abnormalities. The neurological examination showed conflicting findings with mild axial hypotonia and mild spasticity in his extremities, as indicated by accelerated deep tendon reflexes. The auditory brainstem response clearly showed all five response peaks, but a mild deafness pattern was indicated by the fact that the response could be recorded over 50 dB. Almost all laboratory examinations showed no abnormalities, but thyroid functions showed abnormal thyroid function with a high TSH level of 5.93 μg/ml (normal range: 0.49–4.94), a high free T3 level of 6.37 pg/ml (normal range: 1.71–3.71) and a normal free T4 level of 0.75 ng/dl (normal range: 0.7–1.48). A brain MRI examined at the age of 8 months showed no marked abnormality, but subsequent analysis at 17 months showed a delayed myelination pattern and mild dilatation of the extra-cerebrum space (Figure 1c, d). Chromosomal examination showed a normal male karyotype of 46,XY. At present, the patient shows severe developmental delay with no head control and no turning over. He is declared bedridden.
We suspected that SLC16A2 mutations would be related in these two patients because of the combined presentation of delayed myelination and thyroid dysfunction with an elevated T3 pattern. After obtaining written informed consent, blood samples were obtained from both families for the purpose of molecular diagnosis. This study was approved by the ethical committee in Tokyo Women’s Medical University. Genomic DNA extracted using a QIAamp DNA extraction kit (Qiagen, Hilden, Germany) was used for PCR-based direct Sanger sequencing of SLC16A2.
In patient 1, a 3-bp deletion (c.1390_1392delCCC), leading to an in-frame amino-acid deletion (P464del), was identified in exon 5 (Figure 2a). This deletion has never been previously reported. Because his mother declined to be genotyped, we do not know whether this mutation is de novo or familial.

(a) c.1390_1392delCCC is shown in SLC16A2 exon 5 in patient 1. (b) Patient 2 and his mother are hemizygous and homozygous for c.97T>C in SLC16A2 exon 1 (S33P), respectively. (c) Patient 2 and his mother are hemizygous and heterozygous for c.449C>T in SLC16A2 exon 2 (A150V).
In patient 2, we identified two single-nucleotide variants (SNVs) that lead to amino-acid changes: c.97T>C in exon 1 (S33P) and c.449C>T in exon 2 (A150V) (Figure 2b,c). The SNVs were listed in dbSNP build 138 as rs6647476 and rs104894936, respectively. The damaging effect scores of the SNVs were calculated using PolyPhen-2 and SIFT; however, no difference was observed between the SNVs. The PolyPhen-2 scores were 0.898 (S33P) and 0.673 (A150V), indicating that they were benign. SIFT scores were 0.47 (S33P) and 0.25 (A150V), indicating that the SNVs were TOLERATED. The minor allele frequency (MAF) of S33P was 36.632%, indicating that it is a common SNP with ⩾1% MAF. In comparison, the MAF of A150V is low, at <1%, indicating that it is a flagged SNP. Based on these data, S33P was considered benign, whereas A150V was considered pathogenic. The mother of patient 2 was homozygous and heterozygous for S33P and A150V, respectively, showing that these variants co-segregated in the maternally derived allele.
In the present study, we diagnosed two patients with AHDS who showed severe developmental delay, especially with respect to motor development. Neither patient acquired head control. The results of the thyroid function examination showed elevated T3 levels in both patients despite variable TSH and T4 levels. This is the typical pattern for AHDS. Brain MRI showed severely delayed myelination in both patients. Additionally, the total brain volume was mildly reduced in both patients.
Molecular analysis identified a novel single-amino-acid deletion arising from a 3-bp nucleotide deletion in patient 1 (P464del). A missense mutation, P464L, affecting this residue had been previously reported.6 A150V identified in patient 2 has been recurrently reported,7–9 and functional analysis of this mutation confirmed that it results in a complete loss of specific T3 uptake8 and damaged T3 metabolism.10 To our knowledge, co-segregation of S33P and A150V was reported for the first time in the present study. Although we had no evidence of whether A150V was inherited in this family or occurred de novo, the relatively frequent occurrence of A150V in AHDS patients suggests that this region containing a “CGCG” repeat sequence may be a mutation hot spot.
In conclusion, we identified an in-frame/single-amino-acid deletion as well as a recurrent missense mutation in AHDS patients. Because both patients showed similar clinical manifestations, no clear genotype–phenotype correlation was observed for SLC16A2 alterations.
References
References
Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff SA . A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet 2004; 74: 168–175.
Heuer H, Maier MK, Iden S, Mittag J, Friesema EC, Visser TJ et al. The monocarboxylate transporter 8 linked to human psychomotor retardation is highly expressed in thyroid hormone-sensitive neuron populations. Endocrinology 2005; 146: 1701–1706.
Vaurs-Barriere C, Deville M, Sarret C, Giraud G, Des Portes V, Prats-Vinas JM et al. Pelizaeus-Merzbacher-Like disease presentation of MCT8 mutated male subjects. Ann Neurol 2009; 65: 114–118.
Gika AD, Siddiqui A, Hulse AJ, Edward S, Fallon P, McEntagart ME et al. White matter abnormalities and dystonic motor disorder associated with mutations in the SLC16A2 gene. Dev Med Child Neurol 2010; 52: 475–482.
Friesema EC, Visser WE, Visser TJ . Genetics and phenomics of thyroid hormone transport by MCT8. Mol Cell Endocrinol 2010; 322: 107–113.
Papadimitriou A, Dumitrescu AM, Papavasiliou A, Fretzayas A, Nicolaidou P, Refetoff S . A novel monocarboxylate transporter 8 gene mutation as a cause of severe neonatal hypotonia and developmental delay. Pediatrics 2008; 121: e199–e202.
Friesema EC, Grueters A, Krude H, von Moers A, Reeser M et al. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet 2004; 364: 1435–1437.
Biebermann H, Ambrugger P, Tarnow P, von Moers A, Schweizer U, Grueters A . Extended clinical phenotype, endocrine investigations and functional studies of a loss-of-function mutation A150V in the thyroid hormone specific transporter MCT8. Eur J Endocrinol 2005; 153: 359–366.
Jansen J, Friesema EC, Kester MH, Schwartz CE, Visser TJ. Genotype-phenotype relationship in patients with mutations in thyroid hormone transporter MCT8. Endocrinology 2008; 149: 2184–2190.
Jansen J, Friesema EC, Kester MH, Milici C, Reeser M, Gruters A et al. Functional analysis of monocarboxylate transporter 8 mutations identified in patients with X-linked psychomotor retardation and elevated serum triiodothyronine. J Clin Endocrinol Metab 2007; 92: 2378–2381.
Data Citations
Yamamoto, Toshiyuki HGV Database (2014) http://dx.doi.org/10.6084/m9.figshare.hgv.507
Yamamoto, Toshiyuki HGV Database (2014) http://dx.doi.org/10.6084/m9.figshare.hgv.509
Yamamoto, Toshiyuki HGV Database (2014) http://dx.doi.org/10.6084/m9.figshare.hgv.511
Acknowledgements
We would like to express our gratitude to the patients and their families for their cooperation. This work was partially supported by a Grant-in-Aid for Scientific Research from Health Labor Sciences Research Grants from the Ministry of Health, Labor, and Welfare, Japan (TY).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Yamamoto, T., Shimojima, K., Umemura, A. et al. SLC16A2 mutations in two Japanese patients with Allan–Herndon–Dudley syndrome. Hum Genome Var 1, 14010 (2014). https://doi.org/10.1038/hgv.2014.10
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2014.10
