
Abstract
Chromosomal rearrangements such as inversions can play a crucial role in maintaining polymorphism underlying complex traits and contribute to the process of speciation. In Atlantic cod (Gadus morhua), inversions of several megabases have been identified that dominate genomic differentiation between migratory and nonmigratory ecotypes in the Northeast Atlantic. Here, we show that the same genomic regions display elevated divergence and contribute to ecotype divergence in the Northwest Atlantic as well. The occurrence of these inversions on both sides of the Atlantic Ocean reveals a common evolutionary origin, predating the >100 000-year-old trans-Atlantic separation of Atlantic cod. The long-term persistence of these inversions indicates that they are maintained by selection, possibly facilitated by coevolution of genes underlying complex traits. Our data suggest that migratory behaviour is derived from more stationary, ancestral ecotypes. Overall, we identify several large genomic regions—each containing hundreds of genes—likely involved in the maintenance of genomic divergence in Atlantic cod on both sides of the Atlantic Ocean.
Similar content being viewed by others


Introduction
Genomic islands of divergence (Wu, 2001; Nosil et al., 2009) are known to emerge through divergence hitchhiking (Via, 2012) but can also evolve through other processes that reduce recombination in genomic regions, such as inversions (Kirkpatrick and Barton, 2006). Chromosomal rearrangements in the form of inversions have been hypothesized to play a key role in maintaining polymorphism in complex traits (Conrad and Hurles, 2007). Within inversions, the rate of crossing over is reduced by several orders of magnitude, allowing genomic islands of divergence within inversions to be larger than in collinear regions. If an inversion captures several locally adapted alleles, it can be integral to the genomic process of local adaptation because it suppresses meiotic recombination in heterozygous individuals (Kirkpatrick and Barton, 2006).
Inversion polymorphisms have been linked to adaptation with gene flow in several species including Drosophila (Noor et al., 2001), Helianthus sunflowers (Rieseberg, 2001), Anopheles mosquitoes (Ayala and Coluzzi, 2005) and Agrodiaetus butterflies (Kandul et al., 2007). Recently, evidence of genomic islands of divergence caused by chromosomal inversions has been reported in several studies focussing on Atlantic cod (Gadus morhua L.) (Berg et al., 2016; Sodeland et al., 2016; Kirubakaran et al., 2016; Barth et al., 2017). Inversions that differentiate migratory from nonmigratory ecotypes (Berg et al., 2016; Kirubakaran et al., 2016) have been documented for cod in the Northeast Atlantic, either in only a small fraction of the genome (Kirubakaran et al., 2016) or a few distinct populations (Berg et al., 2016; Sodeland et al., 2016). Existence of these inversions in the Northwest Atlantic or around Iceland has not yet been investigated, although genomic islands of divergence have previously been documented for several populations (Bradbury et al., 2010, 2013; Hemmer-Hansen et al., 2013; Berg et al., 2015, 2016). The fact that such ‘islands’ have previously been identified on both sides of the Atlantic (Bradbury et al., 2010, 2013) suggests that inversions might also play a role in explaining genomic islands of divergence in Northwest Atlantic and Icelandic cod. Interestingly, the allele frequencies of these ‘islands’ display parallel latitudinal clines in populations that are otherwise genetically distinct, on both sides of the Atlantic (Bradbury et al., 2010), that is indicative of parallel evolution. Furthermore, a subset of the single-nucleotide polymorphisms (SNPs) investigated by Bradbury et al. (2010) has also been associated with temperature in several other studies (Nielsen et al., 2009; Hemmer-Hansen et al., 2013; Therkildsen et al., 2013).
Here, we investigate Atlantic cod populations from both sides of the Atlantic Ocean (Table 1) that have previously been partitioned into (1) a northern (Can-N) and a southern (Can-S) group (Bradbury et al., 2010, 2013) in the Northwest Atlantic; (2) Frontal and Coastal ecotypes in Iceland (see, for example, Pampoulie et al., 2008, 2015); and (3) migratory North East Arctic cod (NEAC) and nonmigratory Norwegian coastal cod (NCC) in the Northeast Atlantic. First, we identify outlier SNPs and genomic regions putatively under selection for each population pair and between the spatially different subset of populations within each of the three broadly delineated groups (comparing Can-N with Can-S, Frontal with Coastal and NEAC with NCC), and then make a trans-Atlantic comparison of the observed genomic patterns. Second, we explore linkage disequilibrium (LD) patterns, combined with SNP information, to look for chromosomal rearrangements and to investigate whether previously identified inversions (Berg et al., 2016; Sodeland et al., 2016; Kirubakaran et al., 2016) occur throughout the distribution range of Atlantic cod. Finally, we investigate the frequencies of the inversions among populations, both within and across the continents, to unravel their distributions and likely origins, and discuss possible mechanisms driving the observed patterns in the light of ecotype divergence and adaptation. The results provide insight into the process of genomic divergence in marine fishes in general.
Materials and methods
Samples, DNA extraction and genotyping
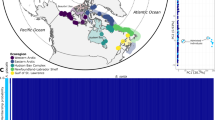
We sampled 316 Atlantic cod (Figure 1 and Table 1), consisting of 144 individuals from 5 locations from the Northwest Atlantic, 39 Frontal and 39 Coastal ecotype individuals from Iceland (classified by data storage tag (DST) profiles, see Pálsson and Thorsteinsson, 2003; Thorsteinsson et al., 2012), and 50 NEAC and 44 NCC individuals from the Northeast Atlantic.

Map showing the sampling locations of the Atlantic cod populations used in the present study. With one exception, red dots indicate the position where the samples were collected; the Icelandic samples were collected at several locations in the waters around Iceland and later categorized as Frontal or Coastal, based on Data Storage Tag profiling. See Figure 1 in Thorsteinsson et al. (2012) for a detailed view of the Icelandic sampling localities and see Table 1 for a detailed description of the populations in the present study. Can-N_PB, Placentia Bay; Can-N_SG, Southern Gulf of St Lawrence; Can-S_SB, Sambro; Can-S_GM, Gulf of Maine; Can-S_BB, Browns Bank; Ice_F, Iceland Frontal; Ice_C, Iceland Coastal; NEAC, Northeast Arctic cod; NCC, Norwegian coastal cod. The map was modified from http://www.graphic-flash-sources.com/world-vector-map/ using Adobe Illustrator CC.
DNA was extracted from muscle tissue using the E.Z.N.A Tissue DNA kit (Omega Bio-Tek, Norcross, GA, USA) and normalized to 100 ng μl−1. All samples were individually genotyped using a 12K Illumina SNP chip for which 8165 SNPs were polymorphic in this data set, had a call rate of >95% and showed Mendelian inheritance in a separate set of individuals with a pedigree. Out of these SNPs, 602 were close to selected candidate genes, 1470 were nonsynonymous SNPs and the remaining 5857 SNPs were randomly distributed throughout the 23 different linkage groups (LGs). Genotype clustering was performed in Genome Studio 2011.1 (Illumina Inc., San Diego, CA, USA). The nomenclature of the LGs follows Hubert et al. (2010) and the order of the SNPs are as in Berg et al. (2016). All 8165 SNPs used were mapped to the published Atlantic cod genome (ATLCOD1C) (Star et al., 2011) in the same way as in Berg et al. (2016) and details are available in dbSNP (www.ncbi.nlm.nih.gov/snp).
Outlier detection and population genetics
Allele frequencies and observed and expected heterozygosity (Ho and He) within each population were calculated in ARLEQUIN 3.5.1.3 (Excoffier and Lischer, 2010). Departure from Hardy–Weinberg equilibrium was tested locus by locus in each population in ARLEQUIN with 100 000 iterations and a Markov Chain of 1 000 000. Correction for multiple testing was performed in R (R Core Team, 2012), using the QVALUE package (Storey, 2002) with a q-value of 0.05 as a threshold.
Outlier detection in the respective data sets was performed using 10 independent runs of BAYESCAN v2.1 (Foll and Gaggiotti, 2008), using stringent criteria, assuming selection to be 10% and false discovery rate set to 0.01. We report both the median log10(posterior odds) and the median q-value. As outlier tests may have a high rate of false positives because of the effects of population demography and bottlenecks (Narum and Hess, 2011; de Villemereuil et al., 2014), and because of the clear trans-Atlantic divergence in the data, we performed outlier analyses pairwise between all population pairs or identified groups to reduce the methodological weakness caused by population structuring (Vitalis et al., 2001).
Based on the outlier analyses, SNPs were categorized as outliers or as neutral. To avoid bias in the FST and STRUCTURE (Pritchard et al., 2000) analyses, tag SNPs based on LD values between SNPs (r2>0.5) were selected using PLINK v1.07 (Purcell et al., 2006). The outlier and neutral data sets (518 and 7369 SNPs) are represented by 325 and 7075 unlinked tag-SNPs. Locus-specific FST values and weighted average FST values between all populations were calculated in ARLEQUIN, using 10 000 permutations. We calculated nucleotide diversity (π) within all identified groups, and nucleotide divergence between these groups (DXY), using a sliding windows approach with a 50-SNP window and 10 SNPs per iteration in DnaSP 5.10 (Librado and Rozas, 2009). These analyses were also performed locally within each of the identified chromosomal rearrangements.
Discriminant analysis of principal components (DAPC), using all 8165 SNPs were performed, using the R package ADEGENET (Jombart and Ahmed, 2011). The correlated allele frequency and admixture model in STRUCTURE was used to identify major genetic clusters in the data set, performing 10 independent runs for each value of K (burn-in of 10 000 Markov chain Monte Carlo iterations followed by 100 000 iterations) on the different data sets. Delta K and the best K-value for each data set in STRUCTURE was identified with CLUMPAK (Kopelman et al., 2015). NETVIEW P (Steinig et al., 2016) was used to visualize the neutral population divergence in the data based on an isolation by state (1−IBS) matrix constructed in PLINK at k=50, using 7075 unlinked neutral SNPs. The network construction is independent of prior population information and based solely on the genetic distance between individuals.
LD and rearrangement patterns
The presence of intrachromosomal LD, quantified with the r2 estimate using PLINK, was evaluated in all populations separately and within the identified groups (Table 1). The R package inveRsion (Cáceres et al., 2012) was used to detect and locate potentially inverted genomic regions and to identify the inversion status of each individual, using block size=3, min. allele=0.1 and thbic=0. This method utilizes the LD differences across inversion breakpoints to detect potentially inverted regions. To complement this LD-based approach, the R package invClust (Cáceres and González, 2015) was also used to identify potentially inverted regions by haplotype tagging and dimensionality reduction analysis based on predefined regions of interest. These regions were defined based on the LD analyses performed in PLINK. DAPCs were performed within the identified inversions to visualize the distinct three-cluster pattern, reflecting the different inversion genotypes. Simulation analyses have demonstrated that such analyses can be used efficiently to detect and genotype inversion polymorphisms of unphased SNP data (Ma and Amos, 2012).
Results
We investigated a total of 8165 SNPs, distributed throughout 23 LGs with an average distance of 94 000 bp between SNPs, based on a genome size of 830 Mb (Star et al., 2011), in 316 individuals of cod from both sides of the Atlantic Ocean (Figure 1 and Table 1). A total of 5202 SNPs were located within 5000 bp of 4245 Ensembl annotated genes. Only seven SNP loci were significantly out of Hardy–Weinberg Equilibrium, after false discovery rate correction (q<0.05), in any of the populations (Supplementary Table S1), indicating no Wahlund effect. The number of polymorphic loci and the observed and expected heterozygosity was generally lower in Northwest Atlantic populations than in Northeast Atlantic populations (Table 1).
Population divergence
Neutral weighted FST between the Northwest and Northeast Atlantic was 0.081 and all pairwise FST values were significantly different from zero except for the Can-S_BB comparisons with Can-S_SB and Can-S_GM and the Ice_F/Ice_C comparison (Supplementary Table S2; see Table 1 for sample codes). The FST values based on the outlier SNPs (see below) were generally orders of magnitude larger than those based on the neutral SNPs in any pairwise comparison and only the Can-S_BB comparisons with Can-S_SB and Can-S_GM were not significantly different from zero. Elevated FST values predominantly occurred within distinct regions in LGs 2, 7 and 12 (but also to some extent in LG1) in the Northwest Atlantic populations, primarily in LGs 1, 2 and 7 in the Northeast Atlantic populations, and in a distinct region in LG23 and in a few SNPs in LG11 between the two continents (Supplementary Figure S1). This pattern corresponds well with the distinctly different heterozygosity and nucleotide divergence (DXY) patterns observed (Supplementary Figures S2 and S3).
Bayesian cluster analyses as implemented in STRUCTURE supported a distinct separation (ΔK=2) between Northwest and Northeast Atlantic populations, using both the neutral and the outlier data sets (Supplementary Figure S4). In addition, the STRUCTURE analysis based on the neutral data set (K=3) revealed that the NCC population is distinctly different from the other Northeast Atlantic populations. Further neutral population structuring within both the Northwest and Northeast Atlantic was evident from the network analyses (Figure 2). The DAPC, using all SNPs, confirmed the distinct separation between Northwest and Northeast Atlantic populations (Figure 3a), and also revealed a further stratification within these regions (Figures 3b and d). Within the Northwest Atlantic, the Can-S_SB clustered together with the Can-S populations that clustered separately from the Can-N populations (Figure 3b). Within the Northeast Atlantic, the Frontal and Coastal ecotypes from Iceland clustered closer to the migratory NEAC population, whereas the nonmigratory NCC population was distinctly different from these. When grouping the populations into Can-N/migratory and Can-S/non-migratory entities (Table 1), we observed clear genomic differences that could be attributed to the putative inversions within LGs 1, 2, 7 and 12 (Supplementary Figures S5a and b). Moreover, the separation pattern within the different LGs primary reflected the frequency differences between these regions (Figures 3c and e, Supplementary Figures S5c–f). The remaining LGs showed little differentiation between these two groups (Supplementary Figure S5g).

Neutral population divergence between Atlantic cod populations. Population clustering based on 7075 neutral SNPs in 316 individuals of Atlantic cod, using an isolation-by-state (IBS) matrix constructed in PLINK, visualized using the NETVIEW P pipeline at k=50, capturing large-scale genetic differentiation across the Atlantic as well as fine-scale structuring within the Northwest and Northeast Atlantic populations. Edge width is proportional to the genetic distance between individuals. Can-N_PB, Placentia Bay; Can-N_SG, Southern Gulf of St Lawrence; Can-S_SB, Sambro; Can-S_GM, Gulf of Maine; Can-S_BB, Browns Bank; Ice_F, Iceland Frontal; Ice_C, Iceland Coastal; NEAC, Northeast Arctic cod; NCC, Norwegian coastal cod.

Spatial relationship between and within Northwest and Northeast Atlantic cod based on discriminant analysis of principal components (DAPC). Based on all 8165 SNPs, a distinct trans-Atlantic separation and a clear separation within Northeast and Northwest Atlantic is observed (a). The stratification within the Northwest Atlantic (b) and the Northeast Atlantic (d) is even more evident when these groups are analysed separately. The loading plots based on the DAPC analyses show the contribution of each SNP to the differentiation within the Northwest Atlantic (c) and Northeast Atlantic (e) populations. The analyses are based on n.pca=3 and n.da=2, calculated in ADEGENET that assumes a predefined population designation of the individuals. Can-N_PB, Placentia Bay; Can-N_SG, Southern Gulf of St Lawrence; Can-S_SB, Sambro; Can-S_GM, Gulf of Maine; Can-S_BB, Browns Bank; Ice_F, Iceland Frontal; Ice_C, Iceland Coastal; NEAC, Northeast Arctic cod; NCC, Norwegian coastal cod.
Outlier detection and identification of genomic regions under selection
Outlier analyses were performed pairwise, and identified 227 SNPs (2.8%) as candidates for divergent selection (q<0.01) in the Northwest Atlantic populations, 361 SNPs (4.4%) in the Northeast Atlantic, and 518 SNPs (6.3%) in all pairwise population comparisons (Supplementary Figure S6 and Supplementary Table S3). Outlier tests were also performed between the Can-N and Can-S groups, between the migratory and nonmigratory groups and between the Can-N/migratory and Can-S/nonmigratory entities, and identified 237, 319 and 365 SNPs as candidates for divergent selection, respectively (Figure 4 and Supplementary Table S3). The outlier analyses revealed four large regions potentially under selection in LGs 1, 2, 7 and 12 (Figure 4 and Supplementary Figure S6) consisting of 170, 47, 162 and 75 SNPs, respectively. The outlier regions in LGs 2 and 7 were present on both sides of the Atlantic: the outlier region in LG1 was predominantly present in Northeast Atlantic comparisons and the region in LG12 was only divergent in the Northwest Atlantic comparisons. A few additional nonlinked outliers were detected in all but two LGs (Supplementary Table S3). Of the 518 outlier SNPs, 364 are located either in or within 5 kb of a known gene, of which 196 are located in exons and 161 are nonsynonymous substitutions (Supplementary Table S3).

Manhattan plots visualizing the pairwise outlier patterns between ecotypes of Atlantic cod. The observed outlier pattern between Can-N and Can-S (a) indicates that the majority of outliers are clustered within the inversions in LGs 2, 7 and 12, whereas the inversions within LGs 1, 2 and 7 are putatively under selection between migratory and nonmigratory ecotypes in the Northeast Atlantic (b). By grouping all individuals into migratory and nonmigratory groups (see Table 1 for details), outliers are detected within the inversions in LGs 1, 2, 7 and 12 (c). The plots are based on median log10 posterior odds (PO) values from 10 independent runs of BAYESCAN. SNPs are plotted according to linkage group and position within the linkage groups along the x axis as in Berg et al. (2016). The red line at 1 indicates ‘strong association’ according to Jeffreys (1961). The migratory group consists of Can-N_PB (Placentia Bay), Can-N_SG (Southern Gulf of St Lawrence), Ice_F (Iceland Frontal) and NEAC (Northeast Arctic cod), whereas the nonmigratory group consists of Can-S_SB (Sambro), Can-S_GM (Gulf of Maine), Can-S_BB (Browns Bank), Ice_C (Iceland Coastal) and NCC (Norwegian coastal cod). For visualization purposes, maximum log10 (PO) values are set to 5 (all underlying values are found in Supplementary Table S3).
LD patterns and chromosomal rearrangements
In LGs 1, 2, 7 and 12, a substantial number of SNPs were detected in high LD, forming distinct LD blocks (Supplementary Figure S7a). The strength of LD within LG1 differed between Northwest and Northeast Atlantic populations, and also between the different groups; it was greater in the Northeast Atlantic migratory populations than in the Northwest Atlantic Can-S populations (Supplementary Figure S7b). In LGs 2 and 7, the LD patterns were similar in all groups, except for the Can-N population in the Northwest Atlantic, where LD was consistently low (Supplementary Figure S7b). Similarly, in LG12, differences were observed between the Northwest and Northeast Atlantic populations, with distinctly low LD within the Can-N populations (Supplementary Figure S7b). The LD analyses also revealed smaller regions of high LD in other LGs (Supplementary Figure S7a).
By using the R packages InveRsion and InvClust, the linked regions under selection in LGs 1, 2, 7 and 12 were identified as putative inversions, in addition to a potential inversion not under selection in LG23 (Table 2). The inversion breakpoints identified by the InveRsion package correspond well with the identified boundaries for the blocks in high LD (Supplementary Table S4). The different genotypic combinations (inversion frequencies) at LGs 1, 2, 7 and 12 (Supplementary Table S5) contribute to the observed population and ecotype divergence in addition to a trans-Atlantic difference (Table 2 and Figure 5), whereas the LG23 region primarily shows a trans-Atlantic difference. FST values within the homozygote inverted and noninverted variants were calculated for all four divergent regions across the Atlantic Ocean (Table 2), showing large increased diversity in the noninverted variant in LG1 and LG12 and in the inverted variant in LG7, relative to the genome-wide neutral divergence. These results are consistent with the spatial distribution visualized in Figure 5. In addition, low genomic divergence, reflected by heterozygosity (Supplementary Figure S2) and nucleotide diversity (π, Table 2 and Supplementary Figure S3) was observed for the migratory and Can-S groups within the divergent regions in LGs 1, 2, 7 and 12.

Discriminant analysis of principal components (DAPC) of the SNPs embedded within the inversions in LGs 1, 2, 7 and 12. Within all LGs a clear trimodal pattern, reflecting the inversion ‘genotypes’, is observed in addition to a broad trans-Atlantic division. Each dot represents an individual; NI/NI and I/I denote the homozygote noninverted and inverted clusters, respectively, whereas the NI/I denotes the heterozygote clusters. Inversion frequencies are listed in Supplementary Table S5. The analyses are based on n.pca=2 and n.da=2, calculated in ADEGENET. Can-N_PB, Placentia Bay; Can-N_SG, Southern Gulf of St Lawrence; Can-S_SB, Sambro; Can-S_GM, Gulf of Maine; Can-S_BB, Browns Bank; Ice_F, Iceland Frontal; Ice_C, Iceland Coastal; NEAC, Northeast Arctic cod; NCC, Norwegian coastal cod.
The inversions vary in size and number of genes: LG1, at least 18.5 Mb and 785 genes; LG2, ∼6 Mb and 293 genes; LG7, at least 10 Mb and 324 genes; LG12, ∼13 Mb and 419 genes; LG23, >3.5 Mb and 97 genes (Supplementary Table S6). Combined, the divergent regions in LGs 1, 2, 7, 12 and 23 are >50 Mb (≈6% of the genome) and contain >1900 genes (Supplementary Table S6).
Discussion
Polymorphic inversions
Ever since seasonal changes in inversion frequencies were observed in Drosophila (Dobzhansky, 1943), the effects of reduced recombination rates within inversions have been linked to adaptation with gene flow, and investigations have shown that sympatric species exhibit more differences caused by inversions than allopatric species (see, for example, Rieseberg, 2001; Noor et al., 2001; Ayala and Coluzzi, 2005; Kandul et al., 2007). Recent research on tropical reef fishes (Martinez et al., 2015) and Estrildid finches (Hooper and Price, 2015) indicate a quicker fixation of inversions in lineages with higher dispersal potential and gene flow, consistent with a theory where gene flow favours diversification of chromosomal rearrangements caused by locally adapted loci (Kirkpatrick and Barton, 2006). In line with these findings, we observe generally low genome-wide divergence interspersed with highly divergent regions (Figure 4) among the investigated Atlantic cod populations, where gene flow could potentially be high because of few physical barriers. In an adaptation with gene flow scenario where different ecotypes are maintained in close proximity and potentially interbreeding, such as in the case of NEAC/NCC and Ice-F/Ice-C, inversion polymorphism (effectively acting as supergenes) could be an important factor in upholding the ecotype diversity. This has been shown in Heliconius butterflies, where supergenes controlling wing mimicry have been attributed to a series of inversions that suppress recombination (Joron et al., 2011; Jones, Salazar, et al., 2012). Chromosomal rearrangements have also been associated with behavioural or ecotype differences in other species such as white-throated sparrow (Zonotrichia albicollis) (Zinzow-Kramer et al., 2015), rainbow trout (Oncorhynchus mykiss) (Pearse et al., 2014) fire ant (Solenopsis invicta) (Wang et al., 2013), stickleback (Gasterosteus aculeatus) (Jones, Grabherr, et al., 2012) and Anopheles mosquitoes (Love et al., 2016). Recently, a haplotype block and a potential inversion associated with different spawning times have been identified in herring (Clupea harengus) across the Atlantic Ocean (Lamichhaney et al., 2017).
In our data set, we observe four distinct large genomic regions of divergence where SNPs are in persistently high LD with each other. These regions are likely chromosomal inversions over several Mb in size (Berg et al., 2016; Kirubakaran et al., 2016) that segregate as biallelic loci within populations. As such, inversions that contain multiple genes involving a certain set of phenotypic traits could be responsible for maintaining vital polymorphisms within the Atlantic cod genome. Alternatively, the observed pattern could also result from secondary contact between previously diverged populations where the inversions form endogenous incompatibilities protected from recombination. In such a scenario, selection would not cause or maintain the inversions per se, but secondary contact could result in genomic homogenization across the genome except for the inverted regions. If so, the inversions would maintain the differentiation by coupling to loci associated with the divergence that is consistent with the coupling hypothesis (Bierne et al., 2011). Nonetheless, we find this explanation less likely as this hypothesis implies fixation of alternative alleles due to genetic incompatibilities, whereas we observe high frequencies of heterozygous individuals at all the inversions. In addition, a large number of F1 hybrids between the Can-N and Can-S groups have been observed (Bradbury et al., 2014) and crosses of breeding stocks of NEAC and NCC are routinely made (Bangera et al., 2015), suggesting a lack of lethal genetic incompatibilities between the potential inversion types.
Regardless of the cause of the selective advantage of the inversions, these islands comprise ‘fixed’ entities that behave like biallelic loci and that appear to resist introgression even though interbreeding in areas of sympatry have been observed (Bradbury et al., 2014). Our results are consistent with the hypothesis that broad-acting selective agents target numerous biological functions. This combined with relatively low level of pairwise genetic divergence throughout the rest of the genome within the Northwest and Northeast Atlantic cod populations suggests that the divergence within the rearrangements is indicative of adaptive divergence. This has also been shown in perennial and annual ecotypes of monkeyflower that differ significantly within an inversion, while high gene flow homogenizes the collinear parts of the genome (Twyford and Friedman, 2015).
Trans-Atlantic genomic divergence
Cod have been extant on both sides of the Atlantic for >100 000 years (Bigg et al., 2008; Carr and Marshall, 2008). Consistent with the expectations of allopatric population differentiation, a distinct separation is observed across the breadth of the species’ range (Figure 3a), concordant with both mitochondrial DNA data (Árnason, 2004; Bigg et al., 2008; Carr and Marshall, 2008) and nuclear DNA data (Bentzen et al., 1996; Pogson, 2001; O'Leary et al., 2007). Demographic processes appear to explain most of the genomic differentiation between the Northwest and Northeast Atlantic populations, although SNPs within a presumptive inversion in LG23 exhibit elevated FST values relative to the genomic average. The putative inversion in LG23 contains at least 97 genes, where two outlier SNPs were detected in a voltage-dependent calcium channel gene (CACNA1S) known to be expressed in early-stage embryos of Danio rerio (Sanhueza et al., 2009). Alternative alleles within this region are nearly fixed between the Northwest and Northeast Atlantic populations (Table 2), with no differentiation within these locations. Low heterozygosity and low nucleotide diversity within the presumed inversion in the Northwest Atlantic populations indicate that this might be the derived variant. The origin of the inversion is likely to pre-date the trans-Atlantic split as both variants have a trans-Atlantic presence (although at low respective frequencies). In addition, there are two distinct population clusters within both the Northwest and the Northeast Atlantic that cannot be attributed to trans-Atlantic divergence. Here, the main genetic differences are attributed to inversions within LGs 1, 2, 7 and 12.
Genomic divergence within the Northeast Atlantic
Even though a distinction between Coastal and Frontal ecotypes in Icelandic waters has been investigated recently (see, for example, Pampoulie et al., 2008; Grabowski et al., 2011; Pampoulie et al., 2015), the genomic basis for these differences has not yet been examined in detail. However, FST patterns in Icelandic waters between coastal and deep-water populations that were not characterized with DSTs (that is, not real migratory vs nonmigratory ecotypes) have been described as reflecting a differentiation similar to that reported between NEAC and NCC populations (Hemmer-Hansen et al., 2013). To date, true Coastal and Frontal ecotypes can only be distinguished by DST profiles (see Pálsson and Thorsteinsson, 2003; Thorsteinsson et al., 2012), although they exhibit different Pan I locus genotypes and differ at the RH1 opsin gene (Pampoulie et al., 2008, 2015), with both genes residing within the LG1 inversion. Our data show that most of the genomic differentiation between the Coastal and Frontal ecotypes (as defined by DSTs) can be attributed to the LG1 inversion, but that smaller FST differences are also observed at LGs 2 and 7. We do not observe any significant neutral divergence (FST=0.0002) between Frontal and Coastal ecotypes, whereas significant nonneutral divergence (mainly within LGs 1, 2 and 7) are observed (FST=0.0547). Both the Frontal and the Coastal ecotypes are inshore cod that spawn at the same spawning grounds and at the same time. The observed pattern with no neutral divergence, and significant nonneutral divergence, is consistent with a divergent selection hypothesis where individuals potentially interbreed at the spawning grounds followed by de novo selection, discriminating the two ecotypes.
Both the Frontal and Coastal ecotypes cluster close to the NEAC population and away from the NCC population (Figure 3d), as indicated by the low neutral divergence between both Coastal and Frontal ecotypes relative to the NEAC (0.0025 and 0.0026) and NCC (0.0062 and 0.0068). Non-neutral divergence between Frontal and Coastal ecotypes are higher than the divergence between Frontal and NEAC (0.0349), but lower than the observed divergence between Frontal and NCC (0.2507) and between Coastal and both NEAC and NCC (0.1610 and 0.1142). Hence, the two Icelandic ecotypes may be derived from NEAC, where local adaptations are forming migratory and nonmigratory ecotypes based on standing genetic variation in the putative inversions in the NEAC genome. As such, the differentiation that we observe between Ice_C and Ice_F, which has been grouped according to behaviour (based on DST tags), probably reflect ecotype divergence in a similar way.
Genomic divergence within the Northwest Atlantic
Within the Northwest Atlantic, the populations cluster into a Can-N and a Can-S group that are known to occupy different thermal regimes (Hutchings et al., 2007; Bradbury et al., 2010, 2013). The northern populations Can-N_PB (Placentia Bay) and Can-N_SG (Southern Gulf of St Lawrence) belong to the ‘cold’ group, the southern populations Can-S_GM (Gulf of Maine) and Can-S_BB (Browns Bank) belong to the ‘warm’ group and the Can-S_SB (Sambro) is located at the transition between the two groups. For the northern populations, tagging experiments show that large individuals of the Can-N_PB population perform relatively long annual migrations (>100 km and up to 500 km) (Lawson and Rose, 2000) with comparatively precise homing to their natal area (Robichaud and Rose, 2011), and the Can-N_SG is known to exhibit even longer annual migration patterns of >225 km for juveniles and up to 650 km for adult fish (Hanson, 1996). In the southern populations, the Can-S_GM population performs limited annual migration within the Gulf of Maine (<65 km) (Ruzzante et al., 1998) and the Can-S_BB are described as being resident to the bank (Zemeckis et al., 2014), where eggs and larvae are likely to be retained by gyres around the bank. Little is currently known about the migration patterns of the Can-S_SB population, but other populations on the Scotian shelf show limited migration patterns (Ruzzante et al., 1998). The northern populations Can-N_PB and the Can-N_SG spawn in the summer months, the southern populations Can-S_GM and Can-S_BB spawn in late winter/early spring and the Can-S_SB population spawns in late fall (Table 1). As such, there are several notable differences between the Can-N and Can-S groups that are not easily disentangled, such as temperature, spawning time and migratory behaviour. It is clear, however, that the divergence between the Can-N and the Can-S ‘ecotypes’ is not only driven mainly by differences in the inversions in LGs 2, 7 and 12 (reflected by both inversion frequencies, FST values and outlier patterns), but also to some extent by differences in the LG1 inversion. This is consistent with results reported by Bradbury et al. (2010) and Hemmer-Hansen et al. (2013) who identified outlier regions primarily within LGs 2, 7 and 12 among Northwest Atlantic populations, but did not attribute this divergence to inversions or to ecotype differentiation. Lately, significant population differentiation has also been observed between spawning groups within the Gulf of Maine and between Georges bank and Gulf of Maine at three large genomic regions in LGs 2, 7 and 12 and increased FST values was observed between spring and winter spawning populations within the LG2 region (Barney et al., 2017).
Population and ecotype differentiation within the divergent regions
The inversion in LG1 is involved in ecotype divergence of Northeast Atlantic populations (Hemmer-Hansen et al., 2013; Berg et al., 2016; Kirubakaran et al., 2016) but shows a less pronounced divergence between Can-N and Can-S in the Northwest Atlantic populations. However, the presumably ancestral (NI/NI) inversion ‘genotype’, found predominantly in the nonmigratory ecotype in the Northeast Atlantic, is found at similarly high frequencies in the Can-S populations. The NI/NI and the I/I variants are highly divergent across the Atlantic (FST=0.237 and 0.159, respectively), relative to the neutral trans-Atlantic divergence (FST=0.081). This suggests local selection pressures acting differently on both of the variant on each side of the Atlantic, but less so for the I/I variant that is associated with migratory behaviour in the Northeast Atlantic. The fact that we detect a similar trans-Atlantic pattern at the LG1 inversion is consistent with Bradbury et al. (2010) that northern samples from the Northwest Atlantic (Davis Strait) and samples from the Barents Sea (presumably NEAC) were not significantly divergent in this region.
SNPs within the presumed inversions in LGs 2 and 7 have previously been linked to temperature (Bradbury et al., 2010), salinity and oxygen levels in the Baltic Sea (Berg et al., 2015), and ecotype divergence of Northeast Atlantic populations (Berg et al., 2016), and are known to be divergent in a wide range of cod populations across the Atlantic (Bradbury et al., 2010; Hemmer-Hansen et al., 2013). Interestingly, within both of these LGs, the Can-N populations are fixed for the I/I variant that is nearly fixed in the migratory ecotypes in the Northeast Atlantic. Notably, there are clear FST differences between the Ice_C and Ice_F ecotypes but these differences are not strong enough to manifest as potential outliers in the outlier tests. Intriguingly, we also observe elevated FST difference in LG7 between the Ice_C (nonmigratory) and NCC populations that may seem inconsistent with the proposed association with migratory behaviour. However, this might reflect the fact that the Ice_C ecotype presumably has a NEAC origin that consists predominantly of the I/I variant. Hence, the standing genomic divergence may not contain sufficient variation-needed for adaptation. Alternatively, the selection pressure on this genomic region might not be strong enough to cause significant differentiation within the selection timeframe.
The presumably inverted genomic region in LG12 has recently been used to discriminate between two Atlantic cod stocks inhabiting the Norwegian Skagerrak coast (Sodeland et al., 2016), and SNPs within this region have been linked to temperature in two separate studies (Bradbury et al., 2010; Berg et al., 2015). This inversion is fixed or nearly fixed for the I/I variant in all populations in our study except for the three Can-S populations that are highly polymorphic. The frequencies of the inversion in LG12 does not differ significantly between the NEAC and NCC populations (Berg et al., 2016), indicating that this inversion may not distinguish migratory from nonmigratory ecotypes per se. Hence, the observed differences between Can-N and Can-S may reflect adaptation to different thermal regimes. Interestingly, the trans-Atlantic divergence at the NI/NI variant (FST=0.736) is high relative to the I/I variant (FST=0.076) and the neutral trans-Atlantic divergence (FST=0.081) that may indicate local selection pressure acting differently on the NI variant in Northeast and Northwest Atlantic.
Identifying the actual targets of selection within inversions or other tightly linked genomic regions is challenging, as recombinations are reduced within inversions causing difficulties in distinguishing true targets of selection from linked false positive signals. We have postulated that both of the Icelandic ecotypes are derived from NEAC. Hence, if the divergence among the Icelandic samples reflects true ecotype divergence, constituted by de novo selection in each generation, the resulting genomic divergence will be based on the standing genetic variation present in the NEAC genome. In NEAC (and Icelandic populations), the I/I variant is almost fixed in LGs 2 and 7 (frequency: 0.98 and 0.94). As a result, almost all variation within the putative LG 2 and 7 inversions will be collinear, allowing for normal recombination to take place. As such, the genomic divergence between Coastal and Frontal ecotypes within these regions may provide valuable insight into the actual targets of selection, because the normal limitations associated with highly linked genomic regions here are omitted. The highest FST values between Coastal and Frontal ecotypes are found in a SNP close to Synaptotagmin (SYT3) in LG2 (FST=0.1152) and in two SNPs (FST=0.1240 and 0.1222) associated with Tyrosinase (TYR), one of which is nonsynonymous in LG7. SYT3 is known to show different expression patterns in resident ‘sneaker’ individuals of Atlantic Salmon (Salmo salar) relative to normal migratory individuals (Aubin-Horth et al., 2005), supporting a potential role in behavioural ecotype divergence. TYR is considered as a clock-controlling gene (Moraes et al., 2014), known to control circadian rhythm of several physiological and behavioural processes (Reppert and Weaver, 2001, 2002). TYR is also involved in regulation of melanin production that influences both skin and retinal pigmentation potentially relevant to vision, depth adaptation and hence vertical migration in fish. This is an interesting finding, as rhodopsin (RH1)—a gene known to mediate dim light vision—is strongly divergent between the two Icelandic ecotypes, indicating an involvement of visual systems in local adaptation of Atlantic cod (Pampoulie et al., 2015).
Combined, our results suggest that: (1) all of the inversion events occurred before the split between Northeast and Northwest Atlantic cod populations, ∼100 000 years ago; (2) the nonmigratory/Can-S group is always dominated by the ancestral collinear inversion genotype (NI/NI) containing the highest nucleotide diversity; (3) nonmigratory behaviour in the Northeast Atlantic appears to be ancestral to migratory behaviour; and (4) inshore Icelandic ecotypes have a presumed NEAC origin. As such, we provide fundamental insight into the evolution of distinct morphs and ecotypes of Atlantic cod with different life-history strategies across the trans-Atlantic barrier. Overall, the data indicate a central role for a few distinct large genomic regions, presumably inversions. The genomic content of these regions are targets of selection, likely to be involved in generating and maintaining adaptive divergence and population differentiation among Atlantic cod throughout its distribution range.
Data archiving
All SNPs are referred to by their ss# or rs# available in dbSNP at www.ncbi.nlm.nih.gov/SNP/. All individual genotype data are available from the Dryad data repository (http://dx.doi.org/10.5061/dryad.b20ps).
References
Aubin-Horth N, Landry CR, Letcher BH, Hofmann HA . (2005). Alternative life histories shape brain gene expression profiles in males of the same population. Proc R Soc B 272: 1655–1662.
Ayala FJ, Coluzzi M . (2005). Chromosome speciation: humans, Drosophila, and mosquitoes. Proc Natl Acad Sci USA 102 (Suppl 1): 6535–6542.
Árnason E . (2004). Mitochondrial cytochrome b DNA variation in the high-fecundity Atlantic cod: trans-Atlantic clines and shallow gene genealogy. Genetics 166: 1871–1885.
Bangera R, Drangsholt T, Nielsen H, Sae-Lim P, Ødegård J, Puvanendran V et al. (2015). Genotype by environment interaction for growth in Atlantic cod (Gadus morhua L.) in four farms of Norway. J Mar Sci Eng 3: 412–427.
Barney BT, Munkholm C, Walt DR, Palumbi SR . (2017). Highly localized divergence within supergenes in Atlantic cod (Gadus morhua within the Gulf of Maine. BMC Genomics 18: 271.
Barth JMI, Berg PR, Jonsson PR, Bonanomi S, Corell H, Hemmer-Hansen J et al. (2017). Genome architecture enables local adaptation of Atlantic cod despite high connectivity. Mol Ecol 26: 4452–4466.
Bentzen P, Taggart CT, Ruzzante DE, Cook D . (1996). Microsatellite polymorphism and the population structure of Atlantic cod (Gadus morhua in the northwest Atlantic. Can J Fish Aquat Sci 53: 2706–2721.
Berg PR, Jentoft S, Star B, Ring KH, Knutsen H, Lien S et al. (2015). Adaptation to low salinity promotes genomic divergence in Atlantic cod (Gadus morhua L.). Genome Biol Evol 7: 1644–1663.
Berg PR, Star B, Pampoulie C, Sodeland M, Barth JMI, Knutsen H et al. (2016). Three chromosomal rearrangements promote genomic divergence between migratory and stationary ecotypes of Atlantic cod. Sci Rep 6: 23246.
Bierne N, Welch J, Loire E, Bonhomme F, David P . (2011). The coupling hypothesis: why genome scans may fail to map local adaptation genes. Mol Ecol 20: 2044–2072.
Bigg GR, Cunningham CW, Ottersen G, Pogson GH, Wadley MR, Williamson P . (2008). Ice-age survival of Atlantic cod: agreement between palaeoecology models and genetics. Proc R Soc B 275: 163–172.
Bradbury IR, Bowman S, Borza T, Snelgrove PVR, Hutchings JA, Berg PR et al. (2014). Long distance linkage disequilibrium and limited hybridization suggest cryptic speciation in Atlantic cod. PLoS ONE 9: e106380.
Bradbury IR, Hubert S, Higgins B, Borza T, Bowman S, Paterson IG et al. (2010). Parallel adaptive evolution of Atlantic cod on both sides of the Atlantic Ocean in response to temperature. Proc R Soc B 277: 3725–3734.
Bradbury IR, Hubert S, Higgins B, Bowman S, Borza T, Paterson IG et al. (2013). Genomic islands of divergence and their consequences for the resolution of spatial structure in an exploited marine fish. Evol Appl 6: 450–461.
Carr SM, Marshall HD . (2008). Intraspecific phylogeographic genomics from multiple complete mtDNA genomes in Atlantic cod (Gadus morhua: origins of the ‘codmother,’ transatlantic vicariance and midglacial population expansion. Genetics 180: 381–389.
Cáceres A, González JR . (2015). Following the footprints of polymorphic inversions on SNP data: from detection to association tests. Nucleic Acids Res 43: e53.
Cáceres A, Sindi SS, Raphael BJ, Cáceres M, González JR . (2012). Identification of polymorphic inversions from genotypes. BMC Bioinformatics 13: 28.
Conrad DF, Hurles ME . (2007). The population genetics of structural variation. Nat Genet 39: S30–S36.
de Villemereuil P, Frichot É, Bazin E, François O, Gaggiotti OE . (2014). Genome scan methods against more complex models: when and how much should we trust them? Mol Ecol 23: 2006–2019.
Dobzhansky T . (1943). Genetics of natural populations IX. Temporal changes in the composition of populations of Drosophila pseudoobscura. Genetics 28: 162–186.
Excoffier L, Lischer HEL . (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Res 10: 564–567.
Foll M, Gaggiotti O . (2008). A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180: 977–993.
Grabowski TB, Thorsteinsson V, McAdam BJ, Marteinsdóttir G . (2011). Evidence of segregated spawning in a single marine fish stock: sympatric divergence of ecotypes in Icelandic cod? PLoS ONE 6: e17528.
Hanson JM . (1996). Seasonal distribution of juvenile Atlantic cod in the southern Gulf of St Lawrence. J Fish Biol 49: 1138–1152.
Hemmer-Hansen J, Nielsen EE, Therkildsen NO, Taylor MI, Ogden R, Geffen AJ et al. (2013). A genomic island linked to ecotype divergence in Atlantic cod. Mol Ecol 22: 2653–2667.
Hooper DM, Price TD . (2015). Rates of karyotypic evolution in Estrildid finches differ between island and continental clades. Evolution 69: 890–903.
Hubert S, Higgins B, Borza T, Bowman S . (2010). Development of a SNP resource and a genetic linkage map for Atlantic cod (Gadus morhua. BMC Genomics 11: 191.
Hutchings JA, Swain DP, Rowe S, Eddington JD, Puvanendran V, Brown JA . (2007). Genetic variation in life-history reaction norms in a marine fish. Proc R Soc B 274: 1693–1699.
Jeffreys H . (1961) Theory of probability 3rd edn Oxford University Press: New York, NY, USA.
Jombart T, Ahmed I . (2011). adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27: 3070–3071.
Jones FC, Grabherr MG, Chan YF, Russell P, Mauceli E, Johnson J et al. (2012). The genomic basis of adaptive evolution in threespine sticklebacks. Nature 484: 55–61.
Jones RT, Salazar PA, ffrench-Constant RH, Jiggins CD, Joron M . (2012). Evolution of a mimicry supergene from a multilocus architecture. Proc Biol Sci 279: 316–325.
Joron M, Frezal L, Jones RT, Chamberlain NL, Lee SF, Haag CR et al. (2011). Chromosomal rearrangements maintain a polymorphic supergene controlling butterfly mimicry. Nature 477: 203–206.
Kandul NP, Lukhtanov VA, Pierce NE . (2007). Karyotypic diversity and speciation in Agrodiaetus butterflies. Evolution 61: 546–559.
Kirkpatrick M, Barton N . (2006). Chromosome inversions, local adaptation and speciation. Genetics 173: 419–434.
Kirubakaran TG, Grove H, Kent MP, Sandve SR, Baranski M, Nome T et al. (2016). Two adjacent inversions maintain genomic differentiation between migratory and stationary ecotypes of Atlantic cod. Mol Ecol 25: 2130–2143.
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I . (2015). CLUMPAK: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Res 15: 1179–1191.
Lamichhaney S, Fuentes-Pardo AP, Rafati N, Ryman N, McCracken GR, Bourne C et al. (2017). Parallel adaptive evolution of geographically distant herring populations on both sides of the North Atlantic Ocean. Proc Natl Acad Sci USA 114: E3452–E3461.
Lawson GL, Rose GA . (2000). Seasonal distribution and movements of coastal cod (Gadus morhua L.) in Placentia Bay, Newfoundland. Fish Res 49: 61–75.
Librado P, Rozas J . (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452.
Love RR, Steele AM, Coulibaly MB, Traore SF, Emrich SJ, Fontaine MC et al. (2016). Chromosomal inversions and ecotypic differentiation in Anopheles gambiae: the perspective from whole‐genome sequencing. Mol Ecol 25: 5889–5906.
Ma J, Amos CI . (2012). Investigation of inversion polymorphisms in the human genome using principal components analysis. PLoS ONE 7: e40224.
Martinez PA, Zurano JP, Amado TF, Penone C, Betancur-R R, Bidau CJ et al. (2015). Chromosomal diversity in tropical reef fishes is related to body size and depth range. Mol Phylogenet Evol 93: 1–4.
Moraes MNdeCM, de Lima LHRG, Ramos BCR, de Oliveira Poletini M, de Lauro Castrucci AM . (2014). Endothelin modulates the circadian expression of non-visual opsins. Gen Comp Endocrinol 205: 279–286.
Narum SR, Hess JE . (2011). Comparison of FST outlier tests for SNP loci under selection. Mol Ecol Res 11 (Suppl 1): 184–194.
Nielsen EE, Hemmer-Hansen J, Poulsen NA, Loeschcke V, Moen T, Johansen T et al. (2009). Genomic signatures of local directional selection in a high gene flow marine organism; the Atlantic cod (Gadus morhua. BMC Evol Biol 9: 276.
Noor MA, Grams KL, Bertucci LA, Reiland J . (2001). Chromosomal inversions and the reproductive isolation of species. Proc Natl Acad Sci USA 98: 12084–12088.
Nosil P, Funk DJ, Ortiz-Barrientos D . (2009). Divergent selection and heterogeneous genomic divergence. Mol Ecol 18: 375–402.
O'Leary DB, Coughlan J, Dillane E, McCarthy TV, Cross TF . (2007). Microsatellite variation in cod Gadus morhua throughout its geographic range. J Fish Biol 70: 310–335.
Pampoulie C, Jakobsdóttir KB, Marteinsdottir G, Thorsteinsson V . (2008). Are vertical behaviour patterns related to the pantophysin locus in the Atlantic cod (Gadus morhua L.)? Behav Genet 38: 76–81.
Pampoulie C, Skirnisdottir S, Star B, Jentoft S, Jonsdottir IG, Hjörleifsson E et al. (2015). Rhodopsin gene polymorphism associated with divergent light environments in Atlantic cod. Behav Genet 45: 236–244.
Pálsson ÓK, Thorsteinsson V . (2003). Migration patterns, ambient temperature, and growth of Icelandic cod (Gadus morhua: evidence from storage tag data. Can J Fish Aquat Sci 60: 1409–1423.
Pearse DE, Miller MR, Abadía-Cardoso A, Garza JC . (2014). Rapid parallel evolution of standing variation in a single, complex, genomic region is associated with life history in steelhead/rainbow trout. Proc R Soc B 281: 20140012.
Pogson GH . (2001). Nucleotide polymorphism and natural selection at the pantophysin (Pan I) locus in the Atlantic cod, Gadus morhua (L.). Genetics 157: 317–330.
Pritchard JK, Stephens M, Donnelly P . (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D et al. (2006). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81: 559–575.
R Core Team. (2012) R: A Language and Environment for Statistical Computing2nd ednR Foundation for Statistical Computing: Vienna, Austria.
Reppert SM, Weaver DR . (2001). Molecular analysis of mammalian circadian rhythms. Annu Rev Physiol 63: 647–676.
Reppert SM, Weaver DR . (2002). Coordination of circadian timing in mammals. Nature 418: 935–941.
Rieseberg LH . (2001). Chromosomal rearrangements and speciation. Trends Ecol Evol 16: 351–358.
Robichaud D, Rose GA . (2011). Multiyear homing of Atlantic cod to a spawning ground. Can J Fish Aquat Sci 58: 2325–2329.
Ruzzante DE, Taggart CT, Cook D . (1998). A nuclear DNA basis for shelf‐ and bank‐scale population structure in northwest Atlantic cod (Gadus morhua: Labrador to Georges Bank. Mol Ecol 7: 1663–1680.
Sanhueza D, Montoya A, Sierralta J, Kukuljan M . (2009). Expression of voltage-activated calcium channels in the early zebrafish embryo. Zygote 17: 131–135.
Sodeland M, Jorde PE, Lien S, Jentoft S, Berg PR, Grove H et al. (2016). ‘Islands of divergence’ in the Atlantic cod genome represent polymorphic chromosomal rearrangements. Genome Biol Evol 8: 1012–1022.
Star B, Nederbragt AJ, Jentoft S, Grimholt U, Malmstrøm M, Gregers TF et al. (2011). The genome sequence of Atlantic cod reveals a unique immune system. Nature 477: 207–210.
Steinig EJ, Neuditschko M, Khatkar MS, Raadsma HW, Zenger KR . (2016). NETVIEW P: a network visualization tool to unravel complex population structure using genome-wide SNPs. Mol Ecol Res 16: 216–227.
Storey JD . (2002). A direct approach to false discovery rates. J R Statist Soc B 64: 479–498.
Therkildsen NO, Hemmer-Hansen J, Hedeholm RB, Wisz MS, Pampoulie C, Meldrup D et al. (2013). Spatiotemporal SNP analysis reveals pronounced biocomplexity at the northern range margin of Atlantic cod Gadus morhua. Evol Appl 6: 690–705.
Thorsteinsson V, Pálsson ÓK, Tómasson GG, Jónsdóttir IG, Pampoulie C . (2012). Consistency in the behaviour types of the Atlantic cod: repeatability, timing of migration and geo-location. Mar Ecol Prog Ser 462: 251–260.
Twyford AD, Friedman J . (2015). Adaptive divergence in the monkey flower Mimulus guttatus is maintained by a chromosomal inversion. Evolution 69: 1476–1486.
Via S . (2012). Divergence hitchhiking and the spread of genomic isolation during ecological speciation-with-gene-flow. Phil Trans R Soc B 367: 451–460.
Vitalis R, Dawson K, Boursot P . (2001). Interpretation of variation across marker loci as evidence of selection. Genetics 158: 1811–1823.
Wang J, Wurm Y, Nipitwattanaphon M, Riba-Grognuz O, Huang Y-C, Shoemaker D et al. (2013). A Y-like social chromosome causes alternative colony organization in fire ants. Nature 493: 664–668.
Wu C-I . (2001). The genic view of the process of speciation. J Evol Biol 14: 851–865.
Zemeckis DR, Martins D, Kerr LA, Cadrin SX . (2014). Stock identification of Atlantic cod (Gadus morhua in US waters: an interdisciplinary approach. ICES J Mar Sci 71: 1490–1506.
Zinzow-Kramer WM, Horton BM, McKee CD, Michaud JM, Tharp GK, Thomas JW et al. (2015). Genes located in a chromosomal inversion are correlated with territorial song in white-throated sparrows. Genes Brain Behav 14: 641–654.
Acknowledgements
We thank Matthew P Kent, Sigbjørn Lien and Mariann Arnyasi at CIGENE, Norwegian University of Life Sciences, for SNP genotyping. The Norwegian Sequencing Centre, University of Oslo, provided initial sequencing for SNP identification. This work is part of the Cod SNP Consortium (CIGENE, CEES, IMR and Nofima) activities. The Research Council of Norway provided funding to KSJ (Grant Numbers 199806 and 187940). We also acknowledge funding from the EU-project CODYSSEY (Q5RS-2002-00813) for the tagging experiment performed by V Thorsteinsson in Icelandic waters and funding from the Natural Sciences and Engineering Research Council of Canada (NSERC) that supported the collection of some Canadian samples.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Berg, P., Star, B., Pampoulie, C. et al. Trans-oceanic genomic divergence of Atlantic cod ecotypes is associated with large inversions. Heredity 119, 418–428 (2017). https://doi.org/10.1038/hdy.2017.54
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2017.54
This article is cited by
-
Cellular profiling of a recently-evolved social behavior in cichlid fishes
Nature Communications (2023)
-
Supergene origin and maintenance in Atlantic cod
Nature Ecology & Evolution (2022)
-
The importance of genomic variation for biodiversity, ecosystems and people
Nature Reviews Genetics (2021)
-
The genomic basis of geographic differentiation and fiber improvement in cultivated cotton
Nature Genetics (2021)
-
Adaptation of Drosophila subobscura chromosomal inversions to climatic variables: the Balkan natural population of Avala
Genetica (2021)
