
Abstract
The domestication of taurine cattle initiated ~10 000 years ago in the Near East from a wild aurochs (Bos primigenius) population followed by their dispersal through migration of agriculturalists to Europe. Although gene flow from wild aurochs still present at the time of this early dispersion is still debated, some of the extant primitive cattle populations are believed to possess the aurochs-like primitive features. In this study, we use genome-wide single nucleotide polymorphisms to assess relationship, admixture patterns and demographic history of an ancient aurochs sample and European cattle populations, several of which have primitive features and are suitable for extensive management. The principal component analysis, the model-based clustering and a distance-based network analysis support previous works suggesting different histories for north-western and southern European cattle. Population admixture analysis indicates a zebu gene flow in the Balkan and Italian Podolic cattle populations. Our analysis supports the previous report of gene flow between British and Irish primitive cattle populations and local aurochs. In addition, we show evidence of aurochs gene flow in the Iberian cattle populations indicating wide geographical distribution of the aurochs. Runs of homozygosity (ROH) reveal that demographic processes like genetic isolation and breed formation have contributed to genomic variations of European cattle populations. The ROH also indicate recent inbreeding in southern European cattle populations. We conclude that in addition to factors such as ancient human migrations, isolation by distance and cross-breeding, gene flow between domestic and wild-cattle populations also has shaped genomic composition of European cattle populations.
Similar content being viewed by others
Introduction
The domestication of taurine cattle (Bos taurus) occurred ~10 000 years ago in the Near East (Götherström et al., 2005), and based on mitochondrial DNA (mtDNA) analysis it is estimated to have incorporated the maternal lineages from around 80 female aurochs (Bos primigenius) in this region (Bollongino et al., 2012). Later on, domesticated cattle became widespread all over Europe through migration of early farmers. As farmers settled in the regions that harboured native European aurochs, sporadic interbreeding might have taken place between domestic cattle and native European aurochs, which persisted in some regions until the Middle Ages. Many previous studies have used mtDNA and Y-chromosome markers to address the hypothesis that local aurochs contributed to the gene pool of modern European domestic cattle. All modern European cattle breeds are characterised by the T mitochondrial haplotype. However, previous studies (Edwards et al., 2007; Scheu et al., 2008) investigating the mitochondrial haplotypes of more than one hundred ancient aurochs samples that were retrieved from multiple sites of northern and central Europe only identified P and E haplotypes, which exclude a significant contribution of local aurochs to the maternal lineages of European domestic cattle. On the other hand, many Italian and one Swiss aurochs not only carried P, but also T mitochondrial haplotypes (Mona et al., 2010; Lari et al., 2011; Schibler et al., 2014), so maternal gene flow from native wild aurochs to local domestic cattle may have taken place there. Contrary to mtDNA, studies involving Y-chromosome haplogroups have remained equivocal as to whether gene flow between local aurochs and domestic cattle has occurred (Götherström et al., 2005; Bollongino et al., 2008). Recently, Park et al. (2015) using genome-wide single nucleotide polymorphisms (SNPs) identified significant enrichment of British aurochs alleles in north European cattle breeds.
Until ~250 years ago, cattle productivity was low compared with the modern standards. Cattle were mainly used in subsistence farming and extensive management, a situation which still persists in much of Africa and South Asia (Felius et al., 2014). Since the Industrial Revolution, an intensification of animal husbandry has led to the development of many specialised breeds with derived traits and uniform appearances. However, several traditional cattle breeds still have retained primitive features of their wild ancestors; such breeds can also be referred to as primitive breeds (Supplementary Material). Previous studies (Tapio et al., 2006; Medugorac et al., 2009) have recognised primitive cattle breeds as a valuable resource for genetic variation. Most of these cattle breeds are hardier than commercial breeds and endure adverse environmental conditions and extensive management with low quality forage better (Sæther et al., 2006). On the other hand, several of these breeds have declining effective population sizes, which erode their genetic diversity (Tapio et al., 2006).
It has been estimated that by 2030, 3–4% of European farmland will be abandoned and converted to nature reserves. Management of such landscapes by grazing cattle is very easy, cheap and environmentally friendly, and requires primitive cattle breeds that only need a minimum of human intervention (Schaich et al. 2010). Thus, investigating past admixture patterns and recent change in demography of primitive cattle breeds is essential to formulate and fulfil the breeding objectives tailored to the purpose of landscape management.
Although economic traits of commercial breeds have been extensively studied (Lu et al. 2014; Raven et al. 2014), the more primitive traits and adaptation to extensive management have not been investigated. Analyses of genome-wide SNP data from aurochs and primitive cattle breeds provide the opportunity to investigate gene flow and admixture processes, which have contributed to the evolution of domestic cattle. Here, we use high-density genome-wide SNP markers to investigate genetic origins, admixture patterns and demographic histories of 27 primitive cattle populations and one British aurochs sample. These populations originate from all over Europe. The Dutch, and the Swiss commercial dairy breeds, as well as the Nelore zebu (B. indicus) are also included for comparing the primitive and highly productive cattle and for assessment of zebu admixture, respectively. We also carry out Runs of homozygosity (ROH)-based analyses as they provide valuable insights into recent demographic changes and breeding practices in livestock (Purfield et al., 2012; Ferenčaković et al. 2013).
Materials and methods
Sample collection and classification
A total of 144 animals from 38 different cattle populations, consisting mainly of primitive cattle breeds (27 breeds) were sampled (Figure 1). Numbers of animals per population varied from 1 to 6 (Supplementary Table S1). The geographic origin of 135 animals was assigned to one of five regions: British and Irish (BRI), Dutch-Baltic (NLD, ancestral to large parts of the Lowland Pied and Baltic Red cattle), Balkan and Italian (BAI, representing Podolian and Busha cattle), Iberian (IBR) and Alpine (ALP, combining the Central Brown and Spotted breed cluster). The remaining nine animals did not belong to any of these groups: Jersey (JE), Heck (HE), Nelore zebu (NE), a Maremmana-Pajuna cross (MP) and British aurochs (AU).

Native origin of sampled cattle breeds used in the population genetic analyses. Colours and shape refer to the geographic region of the breeds in the Europe (light green: British and Irish region breeds, light blue: Dutch region breeds, light yellow: Alpine breeds, light pink: Iberian region breed, lavender: Balkan-Italian breeds and dark yellow: Jersey breed). Breed abbreviations: AL, Alentejana; AN, Angler; AR, Arouquesa; AU, Aurochs; BC, Berrenda en Colorado; BK, Boskarin; BN, Berrenda en Negro; BS, Brown Swiss; BU, Busha; CA, Cardena; CC, Cachea; CH, Chianina; CL, Caldelana; DB, Dutch Belted; DF, Dutch Friesian; EL, English Longhorn; FL, Fleckvieh; GA, Galloway; GW, Groningen Whiteheaded; HE, Heck; HF, Holstein Friesian; HL, Highland; JE, Jersey; KC, Kerry cattle; LI, Lidia; LM, Limia; MA, Maremmana; MI, Mirandesa; MRY, Meuse Rhine Issel; MN, Maronesa; MT, Maltese; NE, Nelore; PA, Pajuna; PO, Podolica; RO, Romenian Grey; SA, Sayaguesa; TU, Tudanca; WP, White Park.
Detail of aurochs specimen
The aurochs humerus bone specimen has been retrieved in 1998 from Carsington Pasture cave in Derbyshire, England and was radiocarbon dated to 6736±68 years BP (Edwards et al. 2010; Park et al., 2015). The mitochondrial sequencing revealed P-haplogroup for the aurochs sample (Edward et al., 2010). Park et al. (2015) extracted DNA and performed the high-throughput sequencing (HTS) using Illumina paired-end DNA sequencing (Illumina, Inc., San Diego, CA, USA). Further information about the aurochs sample and sample processing has been described by Park et al. (2015).
SNP array data
DNA was extracted from hair roots, sperm or blood and genotyped with the Illumina BovineHD Genotyping BeadChip, which contains 777 692 SNPs uniformly spanning the bovine genome. Genotypes were called and processed using the Genome Studio software package (Illumina, Inc.). Samples with a call rate below 90% were excluded from the data set and additional criteria used to filter SNPs from the data set were as follows: (1) monomorphic SNPs; (2) SNPs having more than 5% missing genotypes across all the samples; (3) SNPs located on sex chromosomes of the UMD 3.1 assembly (Zimin et al., 2009); and (4) SNPs showing strand discordances. These quality control steps were carried out using PLINK v1.07 (Purcell et al., 2007). The final data set comprised of 698 452 SNPs for 138 animals.
For the British aurochs sample, 773 659 SNPs, which corresponds to SNP position in Illumina BovineHD Genotyping BeadChip, were extracted from the whole-genome sequencing (WGS) reads. After extracting the 773 659 SNP positions, SNPs were selected for inclusion in the final data set if SNP genotype quality ⩾40 and SNP position read depth ⩾5. After filtering, the final aurochs data set consisted of 562 428 SNPs, of which 544 735 are located on autosomes.
Population genetic structure and admixture
Principal component analysis (PCA) was carried out using the programme SMARTPCA 6.0.1 from the EIGENSOFT package (Patterson et al., 2006). The Reynolds’ genetic distances between breeds with more than two samples were also calculated using the Gendist module of PHYLIP 3.69 (Felsenstein, 1989), which was followed by construction of a NeighborNet graph using SPLITSTREE (Huson, 1998).
Population admixture analysis was carried out after selecting 96 671 SNPs with linkage disequilibrium <0.25 using sliding windows of 50 SNPs with forward shift of 5 SNPs. ADMIXTURE v1.22 (Alexander et al., 2009) was used to carry out the genetic admixture analysis with cross-validation (CV) for values of K ranging from 1 to 40. To test for admixture across primitive cattle populations, we calculated three-population test estimates (f3 statistics, Reich et al., 2009) and their corresponding normalised value (z-scores) using all 698 452 SNPs in the ‘threepop’ module implemented in the TREEMIX software package (Pickrell and Pritchard, 2012). Three-population tests considers triplet of the populations (C; A, B), where C is the test population with A and B as reference populations. We performed all possible triplet combinations. The z-scores were calculated by jack-knifing with blocks of 500 SNPs. If the z-score is significantly negative (z ⩽−3.80, after Bonferroni correction for multiple testing), test population C must have admixture from both the reference populations A and B (Patterson et al., 2012). For test population ‘C’, only breeds with more than one sample were considered.
To assess the direction of gene flow, we calculated D-statistics (Green et al., 2010) as implemented in the ADMIXTOOLS (Patterson et al., 2012) software package. The D-statistics method considers the tree topology (((W, X), Y), Z) where; Z represents the outgroup, Y the source of admixture and W and X are the test populations. The D-statistics method counts the ‘ABBA’ sites, where W and Z share the outgroup allele (A), and X and Y share the derived allele (B) as well as the ‘BABA’ sites, where W and Y shares the derived allele, and X and Z shares the outgroup allele. Admixture between Y and either of the test populations creates a significant difference between the ABBA and BABA counts, with a z-score >3.0 (gene flow between W and Y) or ⩽−3.0 (between X and Y).
For the D-statistics method, we generated a yak (B. grunniens) SNP profile to be used as outgroup by splitting the yak scaffolds (Hu et al., 2012) into 600 bp segments followed by local alignment against bovine genome build UMD 3.1 using bwa-mem (Li, 2013) and calling bovine HD array positions using samtools mpileup (Li et al., 2009). Two approaches were followed to select reads for mpileup. In the first approach, we used all uniquely mapped reads to perform mpileup to maximise the number of allele positions. This approach generated 483 970 SNPs after filtering SNPs missing in either the aurochs or yak sample or both. In the second approach, we only used uniquely mapped reads that completely mapped to the bovine genome to use exclusively high-confidence allele calling. This approach generated 77 558 SNPs after filtering SNPs missing in either the aurochs or yak sample or both. Aurochs or zebu was considered as a possible source of admixture and as test populations we compared either ALP or BAI with all other remaining populations, respectively.
Runs of homozygosity
ROH were detected as described by Purfield et al. (2012) by using PLINK v1.07 with a sliding window of 50 SNPs, allowing <100 kb between two consecutive homozygous SNPs, less than two missing genotypes and one possible heterozygous SNP and a minimum length of 500 kbp.
Results
Population genetic structure and admixture
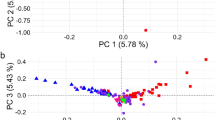
Genetic structure of cattle belonging to various geographical groups was analysed using the PCA and genetic distance-based clustering. The PCA analysis does not take into account breed membership, but yields clear structure as samples from the same population clusters together (Figure 2). The combination of the first two principal components (PC), namely, PC1 and PC2 separates samples according to their geographic origin. PC1 accounts for 16.5% of the total variation and separates B. indicus (Nelore-NE) and B. taurus samples (Figure 2a). PC2 separates the cluster of BRI, NLD (north-western) cattle from another cluster of IBR, ALP and BAI (southern-central) cattle. Furthermore, PC2 positions English Longhorn (EL) samples away from other north-western European cattle samples. To explore the genetic structure only among European cattle, the PCA was performed without B. indicus (NE) and outlier breeds (EL, MT) as identified in the PCA of all samples (Figures 2a and b). Interestingly, the largest PC (PC1) in the PCA of all European samples separates disperse cluster of BAI cattle from the remaining European cattle indicating high divergence of BAI cattle (Figure 2c). PC2 separates densely clustereded IBR cattle from the overlapping cluster of north-western cattle indicating high genotypic similarities among IBR cattle. The PCA analysis also suggests north-western European cattle ancestry for JE breed. The British aurochs sample is positioned in the centre of the plot near the junction of BAI and IBR samples.

PCA plot constructed for PC1 and PC2: (a) on the basis of all available samples; (b) excluding NE samples; (c) excluding MT, EL and NE samples. Samples are grouped based on their geographical region. Abbreviations: ALP, Alpine; AU, Aurochs; BAI, Balkan and Italy; BRI, British and Irish; EL, English Longhorn; MT, Maltese; HE, Heck; IBR, Iberian; JE, Jersey; MP, Maremmana X Pajuna; MT, Maltese; NE, Nelore; NLD, Dutch.
A NeighborNet graph (Supplementary Figure S1) separates breeds according to geographical origin. As expected, outlier breeds suggested by the PCA, such as EL along with Maltese (MT) and Cachena (CC) have branches originating from the centre of the graph. Short branches in the graph also indicate low divergence among Iberian cattle breeds. The IBS distance-based neighbour-joining tree (Supplementary Figure S2), with NE samples assigned as the artificial root, separates samples according to their geographic origin in accordance with the PCA. The BAI population appears to be paraphyletic, whereas IBR, ALP, NLD and BRI formed clear separate clusters.
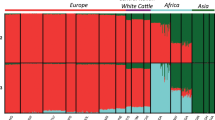
In the ADMIXTURE analysis, estimates of CV decreases with increasing number of user-defined ancestral populations (K) from 1 to 3 (Supplementary Figure S3). Increasing K from 3 to 4 increases the CV only marginally, whereas at K >4, the CV error estimate increases substantially, suggesting K=3 as the most likely number of clusters. The population subdivision at K=2 reproduces the first PCA coordinate by separating NE samples (B. indicus) from all European samples (B. taurus) and also indicates an apparent indicine component in BAI and HE cattle (Figure 3). The population subdivision at K=3 reproduces the second PCA coordinate by separating the IBR cluster from north-western samples. The NLD samples were assigned to a separate cluster at K=4 (data not shown). The single aurochs sample displayed a mosaic of genotypes presents in all clusters estimated across a range of K value from 2 to 8 (data not shown).

ADMIXTURE analysis plot showing model-based population assignments for the values of K=2 (upper) and K=3 (lower). Almost all BAI, AU and HE samples display a possible zebu genetic component at K=2 and K=3. The number below each bar represents the number of samples for the respective population. Abbreviations: ALP, Alpine; BAI, Balkan and Italy; BRI, British and Irish; HE, Heck; IBR, Iberian; JE, Jersey; MP, Maremmana X Pajuna; NE, Nelore; NLD, Dutch. Breed abbreviations: AL, Alentejana; AN, Angler; AR, Arouquesa; BC, Berrenda en Colorado; BK, Boskarin; BN, Berrenda en Negro; BS, Brown Swiss; BU, Busha; CA, Cardena; CC, Cachea; CH, Chianina; CL, Caldela; DB, Dutch Belted; DF, Dutch Friesian; EL, English Longhorn; FL, Fleckvieh; GA, Galloway; GW, Groningen Whiteheaded; HE, Heck; HF, Holstein Friesian; HL, Highland; JE, Jersey; KC, Kerry Cattle; LI, Lidia; LM, Limia; MA, Maremmana; MA, Maltese; ME, Maronesa; MI, Mirandesa; MRY, Meuse Rhine Issel; NE, Nelore; PA, Pajuna; PO, Podolica; RO, Romanian Grey; SA, Sayaguesa; TU, Tudanca; WP, White Park.
Significant negative f3 statistics with NE zebu as one of the reference populations indicates for the BAI breeds (Busha (BU), Chianina (CH) and Maremana (MA)) a zebu ancestry (Supplementary Table S2). A high number of significant negative f3 statistics (Supplementary Table S3) suggests admixture between regional cattle as is observed for all IBR breeds (except Maronesa). The majority of IBR breeds, Chianina (CH), Jersey (JE), White Park (WP), Fleckvieh (FL), Dutch Belted (DB), Dutch Friesian (DF), MRY and Brown Swiss (BS) exhibits evidence of aurochs ancestry (at least one significant f3 test with aurochs as one of the reference populations, Supplementary Table S4). Non-significant f3 statistics indicate isolated breed structures for the majority of the BRI and NLD breeds (Supplementary Table S4).
Contrary to the f3 statistics, D-statistics with yak as outgroup (Z), aurochs as a source of gene flow (Y) and ALP breeds as one of the sister taxa reveals the highest frequencies of the aurochs alleles in the BRI cattle, lower frequencies in the IBR and the NLD cattle and the lowest frequencies in the BAI cattle (Table 1; Supplementary Table S5). In agreement with the previous analyses (PCA, ADMIXTURE and F3), the D-statistics with NE as a source of gene flow (Y) also suggests the highest frequencies of zebu specific alleles in the BAI cattle (Table 2).
Runs of homozygosity
The number of ROH per animal varied from 49 in a Busa (BU) to 357 in a Groningen Whiteheaded (GW) animal. For the total number of ROH per sample per breed (Supplementary Table S6), the EL breed displays the highest mean (~310±14), whereas BU01 cattle displays the lowest mean (~62±14). As expected, the average proportion of the genome coverage by ROH correlates with observed heterozyosity (Ho) (Supplementary Figure S4). The highest average proportion of the genome covered by ROH is observed in isolated breeds with low mean observed heterozygosity: EL (~848±34 Mbp), MT (~825±529 Mbp) and the Iberian Mirandesa (MI, ~783±227 Mbp) (Supplementary Table S6). In two MT animals, the ROH accounted even for a third of their genome. Thus, the ROH coverage in the genome varies considerably within regions with generally high values in a BRI breeds (except WP breed) and a large spread of the value for BAI and IBR populations.
The ROH profiles (size vs number) (Figure 4) display differences in pattern across the different cattle populations. On the regional level, the BRI cattle exhibit the highest average cumulative size as well as the highest average number. On the other hand, many samples from the BAI and IBR cattle display a cumulative size comparable to cattle sampled from the BRI and NLD populations, but with a small number of ROHs, indicating significant contribution primarily from long ROH. Long ROHs in the genome are a consequence of recent inbreeding, whereas short ROHs indicate distant ancestral relatedness such as breed founder effect. To assess the distribution of ROHs length, ROH were further classified into short (0.5–2 Mbp), intermediate (2–8 Mbp) and long (>8 Mbp) (Figure 5). We also note that north-western as well as central European breeds compared with the southern European breeds display more instances of similar ROH patterns for all samples within breed. For instance, all samples of GW breed display a similar distribution of ROH across different categories of ROH length. In fact, for all GW samples more than 95% of the total ROH length belongs to the short to intermediate ROH categories. Individual cattle from the same breed are expected to display similar ROH patterns because of shared population history and similar treatments due to a common herdbook. Comparison of BRI cattle to southern (IBR-BRI) populations shows over representation of all classes of ROH (Figure 5).

The plot displays total number of ROH segments and total ROH size (in Mbp) for all genotyped individuals. Many BAI and IBR samples display a total length comparable to samples from the BRI population. Abbreviations: ALP, Alpine; BAI, Balkan and Italy; BRI, British and Irish; HE, Heck; IBR, Iberian; JE, Jersey; NE, Nelore; NLD, Dutch.

The plot displays breed-wise distribution of ROH segments for each individual in three size classes: (a) short ROH (0.5–2 Mbp); (b) medium ROH (2–8 Mbp) and (c) long ROH (>8 Mbp). Abbreviations: ALP, Alpine; BAI, Balkan and Italy; BRI, British and Irish; IBR, Iberian; NLD, Dutch.
To assess within-breed diversity and effect of selection, exact sharing of ROH haplotypes between animals from the same breed was also assessed (Supplementary Table S7). The NE and EL samples display highest numbers of shared ROHs between two or more individual cattle. Also, BRI (except WP and KC breeds), HF and GW breeds compared with the majority of southern European cattle breeds display higher numbers of shared ROH segments.
Discussion
The inherent ascertainment bias in SNP arrays (Matukumalli et al., 2009) can result in an overestimation of the homozygosity if applied to populations not included in the design of the SNP array. However, the Illumina BovineHD Genotyping BeadChip was designed from a diverse range of breeds comprising zebu, taurine and crossbred individuals. We, therefore, do not expect primitive samples to exhibit a high level of ascertainment bias. Small sample size per breed may distort breed allele frequencies in ADMIXTURE analysis, thus, we only considered the initial K values (from 2 to 4). The validity of D-statistics depends on the number of genetic markers but does not require a large sample size per population (Patterson et al., 2012).
Population origin, admixture and history
The differentiation of indicine and taurine cattle as suggested by PCA and ADMIXTURE is a likely consequence of separate domestication events that occurred within the Neolithic cultures of the Indus Valley and the Fertile Crescent, respectively (Loftus et al. 1994). The PCA and ADMIXTURE analysis also indicated significant genetic differentiation of north-western (BRI and NLD), IBR and BAI cattle populations, which is in agreement with previous findings (Cymbron et al., 2005; Decker et al., 2009; Gautier et al., 2010; Edwards et al., 2011). This north-south difference has been hypothesised to reflect migrations of agriculturalists during the Neolithic transition along the Mediterranean coasts and the Danube river, respectively (Cymbron et al. 2005; Felius et al., 2014).
The divergence of the BAI cattle as suggested by PCA (Figure 2c) can be attributed to an indicine genomic component which is identified in the ADMIXTURE (Figure 3) and D-statistics (Table 2) analyses. By analysing the genome-wide SNP markers, McTavish et al. (2013) and Decker et al. (2014) also reported an indicine influence on Italian cattle breeds. Using whole-genome sequences of ancient human DNA, Jones et al. (2015) and Haak et al. (2015) suggested massive migration of Yamnaya steppe herders as a source of dispersion of Indo-European languages to both northern-central Europe and India. These herders might also have mediated gene flow between Indian zebu and Ukrainian steppe cattle. Contrary to the ADMIXTURE analysis, the three-population tests failed to display evidence for an indicine genetic component in MT and BK, which can be the result of substantial post-admixture drift in these breeds. Investigating population history of Indian population using genome-wide SNPs, Reich et al. (2009) also reported the failure of three-population tests to detect admixture when applied to populations with substantial genetic drift since the admixture.
Given the high genetic diversity in near Eastern cattle populations (Utsunomiya et al., 2014), we expect low ROH abundance in the genomes of BAI cattle due to its proximity to the near East (centre of cattle origin) and presence of an indicine component. The result is in concordance with the expectation, as BAI cattle display low numbers of ROH segments (Figure 4). This pattern of diversity is also seen in humans and pigs, where a relationship exists between ROH abundance and distance to source of origin (Kirin et al., 2010; Bosse et al., 2012). Several BAI breeds such as Busha (BU02), Maltese (MT) and Boskarin (BK), however, display high cumulative ROH length manifested as long ROH (Figure 5c) indicating inbreeding due to recent reduction of the effective population size.
The separate position of the dense IBR cluster in the PCA (Figure 2), short branches in the NJ network (Supplementary Figure S1), and a high number of significant three-population tests (Supplementary Table S4), all suggest a high level of gene flow between IBR cattle breeds, which may have resulted in high genetic similarities and low divergence among IBR breeds. These results probably reflect the geographic isolation of the Iberian Peninsula, and the relatively recent (after 1920) introduction of herd books for Iberian cattle breeds (Felius et al., 2014). Before that time, exchange of breeding material may have been driven more by geographic proximity, resulting in a relatively homogenous gene pool. Analysing BovineHD SNP array data, Cañas-Álvarez et al. (2015) also reported low divergence among Spanish beef cattle breeds, which they attributed to local admixture.
Because of the high admixture among local Iberian cattle identified in the present study and the presence of African cattle ancestry in the genomes of Iberian cattle as reported previously (Decker et al., 2014), we expect low ROH abundance across the genomes of IBR cattle. The result was in concordance with the expectation as IBR cattle display low ROH abundance compared with the BRI cattle. Exceptions were the Cachena (CC) and Mirandesa (MI) (Supplementary Table S6) as well as several individuals of other breeds (Figure 5) with high cumulative ROH length as well as high number of ROH. We propose that this reflects a breed management that does not always avoid crossing of related animals. This may be typical of local populations with a small geographical range. Analysing genetic diversity in Iberian cattle breeds using microsatellite markers, Ginja et al. (2010) also reported low genetic diversity in Mirandesa breed.
The similarity of productive NLD and the more primitive BRI breeds as suggested by the PCA (Figure 2) and ADMIXTURE analysis (Figure 3) may reflect the export of Dutch sires to England since the 16th Century (Felius et al., 2014). The three-population tests (Supplementary Table S4) suggest a well differentiated breed structure for the majority of the BRI and NLD breeds. However, post-admixture genetic drift in the population can mask the signal of the test (Patterson et al., 2012), which is likely in the case of Heck cattle, a complex admixed population derived from several diverse European cattle breeds (Vuure, 2005), in particular, the Hungarian grey, which can explain the minor signature of zebu admixture evident in the ADMIXTURE results. The separate position of the Jersey (JE) breed in the PCA plot (Figure 2c) indicates its virtually complete isolation since 1763 (Felius, 2007).
The fact that cumulative ROH size (Figure 5) is dominated by an abundance of short to intermediate ROHs (0.5–8 Mbp) in NLD as well as BRI breeds indicates the pattern of an isolated population based on a source population of substantial size. An abundance of ROH due to genetic isolation has also been reported in other species such as in Japanese Wild boar (Bosse et al., 2012). The low abundance of long ROH in NLD breeds (Figure 5), particularly in GW and HF breeds, reflects a breed management that is successful in avoiding crossing of related animals probably by maintaining a sufficient effective population size. Isolation of breeds with a relatively small population size increases the probability of inheritance of identical segments as evident clearly in British and Irish region cattle breeds (English Longhorn (EL), HL (Highland) and Galloway (GA)). On the other hand, breeding systems involving intensive culling, usage of few elite bulls and selection in commercial populations may also contribute to increase the overall homozygosity and produce ROH patterns similar to those seen as a result of a bottleneck (Kim et al., 2013). Thus, it is difficult to disentangle the effect of a bottleneck from selection or vice-versa. Hence, high sharing of ROH haplotypes (Supplementary Table S7) as detected between the individuals within BRI breeds can also be the result of confounding process like selection or a bottleneck. The predominant short ROH fragments in EL, HL and KC also indicate a low degree of recent consanguinity. The low genetic diversity of the BRI breeds as observed in terms of high cumulative ROH is in line with previous studies using microsatellite, AFLP and SNP markers (Cymbron et al., 2005; Purfield et al., 2012; Utsunomiya et al., 2014).
Local aurochs gene flow in the gene pool of domestic cattle
The D-statistics (Table 1; Supplementary Table S5) showed the highest frequency of aurochs-derived alleles in BRI cattle. Conversely, the lowest number of aurochs-like derived alleles is reported in BAI cattle. One of the most likely explanations for this result is gene flow from British aurochs (or continental European aurochs from the same meta-population) into the ancestors of north-western and southern-central (IBR-ALP) European cattle breeds. Several scenarios have been put forward for post-domestication hybridisation between wild aurochs and the ancestors of extant domestic cattle breeds (Götherström et al., 2005; Beja-Pereira et al., 2006; Achilli et al., 2008; Schibler et al., 2014). Using 15 498 SNP markers derived from the same aurochs WGS data used here, Park et al. (2015) also provided evidence for gene flow from aurochs into the ancestors of north-western European cattle breeds. Our study, in addition to north-western European breeds, also include various breeds from the Iberian Peninsula and report a high frequency of aurochs-specific-derived alleles in the Iberian cattle compared with the central European and Balkan-Italian cattle. Archaeological findings from the Mesolithic period also indicate a higher concentration of wild aurochs in north-western Europe than southern Europe. A large proportion of aurochs’ fossils has been recovered from Britain indicating high concentration of wild aurochs in Britain during the Mesolithic period (Wright, 2013). In addition, Anderung et al. (2005) also identified P mitochondrial haplotypes in a Bronze Age aurochs sample excavated from northern Spain, indicating a wide geographic distribution of aurochs. On the other hand, the low frequency of aurochs-specific-derived alleles in the BAI cattle may indicates no or very limited contact between the ancestor of the BAI cattle and the British aurochs. Our results, however, do not rule out the possibility of gene flow from different aurochs populations with T haplotypes into the ancestors of the extant BAI cattle.
Further analysis of ancient DNA samples of aurochs and primitive cattle populations from across Eurasia is needed to thoroughly examine aurochs gene flow into ancestors of extant cattle, and to identify the population genomics processes that accompanied domestication. Our data suggest that this is also relevant for further understanding the genetic variation present in European cattle breeds.
Data Archiving
Data available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.f2d1q.
References
Achilli A, Olivieri A, Pellecchia M, Uboldi C, Colli L, Al-Zahery N et al. (2008). Mitochondrial genomes of extinct aurochs survive in domestic cattle. Curr Biol 18: 157–158.
Alexander DH, Novembre J, Lange K . (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Res 19: 1655–1664.
Anderung C, Bouwman A, Persson P, Carretero JM, Ortega AI, Elburg R et al. (2005). Prehistoric contacts over the Straits of Gibraltar indicated by genetic analysis of Iberian Bronze Age cattle. Proc Natl Acad Sci USA 102: 8431–8435.
Beja-Pereira A, Caramelli D, Lalueza-Fox C, Vernesi C, Ferrand N, Casoli A et al. (2006). The origin of European cattle: evidence from modern and ancient DNA. Proc Natl Acad Sci USA 103: 8113–8118.
Bollongino R, Burger J, Powell A, Mashkour M, Vigne JD, Thomas MG . (2012). Modern taurine cattle descended from small number of near-eastern founders. Mol Biol Evol 29: 2101–2104.
Bollongino R, Elsner J, Vigne JD, Burger J . (2008). Y-SNPs do not indicate hybridisation between European Aurochs and domestic cattle. PLoS One 3: 1–5.
Bosse M, Megens HJ, Madsen O, Paudel Y, Frantz LaF, Schook LB et al. (2012). Regions of Homozygosity in the Porcine Genome: Consequence of Demography and the Recombination Landscape. PLoS Genet 8: e1003100.
Cañas-Álvarez JJ, González-Rodríguez A, Munilla S, Varona L, Díaz C, Baro JA et al. (2015). Genetic diversity and divergence among Spanish beef cattle breeds assessed by a bovine high-density SNP chip. J Anim Sci 93: 5164.
Cymbron T, Freeman AR, Isabel Malheiro M, Vigne J-D, Bradley DG . (2005). Microsatellite diversity suggests different histories for Mediterranean and Northern European cattle populations. Proc Biol Sci 272: 1837–1843.
Decker JE, McKay SD, Rolf MM, Kim J, Molina Alcalá A, Sonstegard TS et al. (2014). Worldwide patterns of ancestry, divergence, and admixture in domesticated cattle. PLoS Genet 10: e1004254.
Decker JE, Pires JC, Conant GC, McKay SD, Heaton MP, Chen K et al. (2009). Resolving the evolution of extant and extinct ruminants with high-throughput phylogenomics. Proc Natl Acad Sci USA 106: 18644–18649.
Edwards CJ, Bollongino R, Scheu A, Chamberlain A, Tresset A, Vigne J-D et al. (2007). Mitochondrial DNA analysis shows a Near Eastern Neolithic origin for domestic cattle and no indication of domestication of European aurochs. Proc Biol Sci 274: 1377–1385.
Edwards CJ, Ginja C, Kantanen J, Pérez-Pardal L, Tresset A, Stock F et al. (2011). Dual origins of dairy cattle farming—evidence from a comprehensive survey of european Y-chromosomal variation. PLoS One 6: e15922.
Edwards CJ, Magee Da, Park SDE, McGettigan Pa, Lohan AJ, Murphy A et al. (2010). A complete mitochondrial genome sequence from a mesolithic wild aurochs (Bos primigenius. PLoS One 5: e9255.
Felius M . (2007) Cattle Breeds: An Encyclopedia. Trafalgar Square Books: North Pomfret, VT, USA.
Felius M, Beerling M-L, Buchanan D, Theunissen B, Koolmees P, Lenstra J . (2014). On the history of cattle genetic resources. Diversity 6: 705–750.
Felsenstein J . (1989). PHYLIP-Phylogeny inference package (version 3.2). Cladistics 5: 163–166.
Ferenčaković M, Hamzić E, Gredler B, Solberg TR, Klemetsdal G, Curik I et al. (2013). Estimates of autozygosity derived from runs of homozygosity: empirical evidence from selected cattle populations. J Anim Breed Genet 130: 286–293.
Gautier M, Laloë D, Moazami-Goudarzi K . (2010). Insights into the genetic history of French cattle from dense SNP data on 47 worldwide breeds. PLoS One 5: e13038.
Ginja C, Telo Da Gama L, Penedo MCT . (2010). Analysis of STR markers reveals high genetic structure in portuguese native cattle. J Hered 101: 201–210.
Götherström A, Anderung C, Hellborg L, Elburg R, Smith C, Bradley DG et al. (2005). Cattle domestication in the Near East was followed by hybridization with aurochs bulls in Europe. Proc Biol Sci 272: 2345–2350.
Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M et al. (2010). A Draft Sequence of the Neandertal Genome. Science 328: 710–722.
Haak W, Lazaridis I, Patterson N, Rohland N, Mallick S, Llamas B et al. (2015). Massive migration from the steppe was a source for Indo-European languages in Europe. Nature 522: 207–211.
Hu Q, Ma T, Wang K, Xu T, Liu J, Qiu Q . (2012). The Yak genome database: an integrative database for studying yak biology and high-altitude adaption. BMC Genomics 13: 600.
Huson DH . (1998). SplitsTree: analyzing and visualizing evolutionary data. Bioinformatics 14: 68–73.
Jones ER, Gonzalez-Fortes G, Connell S, Siska V, Eriksson A, Martiniano R et al. (2015). Upper palaeolithic genomes reveal deep roots of modern eurasians. Nat Comm 6: 8912.
Kim E-S, Cole JB, Huson H, Wiggans GR, Van Tassell CP, Crooker Ba et al. (2013). Effect of Artificial Selection on Runs of Homozygosity in U.S. Holstein Cattle. PLoS One 8: e80813.
Kirin M, McQuillan R, Franklin CS, Campbell H, Mckeigue PM, Wilson JF . (2010). Genomic runs of homozygosity record population history and consanguinity. PLoS One 5: e13996.
Lari M, Rizzi E, Mona S, Corti G, Catalano G, Chen K et al. (2011). The complete mitochondrial genome of an 11,450-year-old aurochsen (Bos primigenius) from Central Italy. BMC Evol Biol 11: 32.
Li H . (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997 http://arxiv.org/abs/1303.3997.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al. (2009). The Sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079.
Loftus RT, MacHugh DE, Bradley DG, Sharp PM, Cunningham P . (1994). Evidence for two independent domestications of cattle. Proc Natl Acad Sci USA 91: 2757–2761.
Lu D, Miller S, Sargolzaei M, Kelly M, Vander Voort G, Caldwell T et al. (2014). Genome-wide association analyses for growth and feed efficiency traits in beef cattle. J Anim Sci 91: 3612–3633.
Matukumalli LK, Lawley CT, Schnabel RD, Taylor JF, Allan MF, Heaton MP et al. (2009). Development and characterization of a high density SNP genotyping assay for cattle. PLoS One 4: e5350.
McTavish EJ, Decker JE, Schnabel RD, Taylor JF, Hillis DM . (2013). New World cattle show ancestry from multiple independent domestication events. Proc Natl Acad Sci 110: E1398–E1406.
Medugorac I, Medugorac A, Russ I, Veit-Kensch CE, Taberlet P, Luntz B et al. (2009). Genetic diversity of European cattle breeds highlights the conservation value of traditional unselected breeds with high effective population size. Mol Ecol 18: 3394–3410.
Mona S, Catalano G, Lari M, Larson G, Boscato P, Casoli A et al. (2010). Population dynamic of the extinct European aurochs: genetic evidence of a north-south differentiation pattern and no evidence of post-glacial expansion. BMC Evol Biol 10: 83.
Park SDE, Magee DA, McGettigan PA, Teasdale MD, Edwards CJ, Lohan AJ et al. (2015). Genome sequencing of the extinct Eurasian wild aurochs, Bos primigenius, illuminates the phylogeography and evolution of cattle. Genome Biol 16: 234.
Patterson N, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y et al. (2012). Ancient admixture in human history. Genetics 192: 1065–1093.
Patterson N, Price AL, Reich D . (2006). Population structure and eigenanalysis. PLoS Genet 2: e190.
Pickrell JK, Pritchard JK . (2012). Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet 8: e1002967.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MaR, Bender D et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81: 559–575.
Purfield DC, Berry DP, McParland S, Bradley DG . (2012). Runs of homozygosity and population history in cattle. BMC Genet 13: 70.
Raven L-A, Cocks BG, Hayes BJ . (2014). Multibreed genome wide association can improve precision of mapping causative variants underlying milk production in dairy cattle. BMC Genomics 15: 62.
Reich D, Thangaraj K, Patterson N, Price AL, Singh L . (2009). Reconstructing Indian population history. Nature 461: 489–494.
Sæther NH, B⊘e KE, Vangen O . (2006). Differences in grazing behaviour between a high and a moderate yielding Norwegian dairy cattle breed grazing semi-natural mountain grasslands. Acta Agric Scand Sect A—Anim Sci 56: 91–98.
Schaich H, Rudner M, Konold W . (2010). Short-term impact of river restoration and grazing on floodplain vegetation in Luxembourg. Agric Ecosyst Environ 139: 142–149.
Scheu A, Hartz S, Schmölcke U, Tresset A, Burger J, Bollongino R . (2008). Ancient DNA provides no evidence for independent domestication of cattle in Mesolithic Rosenhof, Northern Germany. J Archaeol Sci 35: 1257–1264.
Schibler J, Elsner J, Schlumbaum A . (2014). Incorporation of aurochs into a cattle herd in Neolithic Europe: single event or breeding? Sci Rep 4: 5798.
Tapio I, Värv S, Bennewitz J, Maleviciute J, Fimland E, Grislis Z et al. (2006). Prioritization for conservation of northern european cattle breeds based on analysis of microsatellite data. Conserv Biol 20: 1768–1779.
Utsunomiya YT, Bomba L, Lucente G, Colli L, Negrini R, Lenstra JA et al. (2014). Revisiting AFLP fingerprinting for an unbiased assessment of genetic structure and differentiation of taurine and zebu cattle. BMC Genet 15: 47.
Vuure VC . (2005) Retracing The Aurochs: History, Morphology & Ecology of An Extinct Wild Ox. Pensoft Pub: Sofia/Moscow.
Wright E . (2013) The History of the European Aurochs (Bos primigenius) from the Middle Pleistocene to its Extinction: An Archaeological Investigation of its Evolution, Morphological Variability and Response to Human Exploitation. University of Sheffield: Sheffield, UK.
Zimin AV, Delcher AL, Florea L, Kelley DR, Schatz MC, Puiu D et al. (2009). A whole-genome assembly of the domestic cow, Bos taurus. Genome Biol 10: R42.
Acknowledgements
MR Upadhyay benefited from a joint grant from the European Commission within the framework of the Erasmus-Mundus joint doctorate ‘EGS-ABG’. We would like to thank Bert Dibbits, Animal Breeding and Genomics Centre, Wageningen University for the DNA extraction and Dr KM (Kyle) Schachtschneider, Animal Breeding and Genomics Centre, Wageningen University for his critical remarks on overall format of manuscript. We would also like to thank Goran Andersson, Department of Animal Breeding and Genetics, Swedish Institute of Agricultural Sciences for his comments on the draft and Professor Mark P Brincat, Department of Obstetrics and Gynaecology, Mater Dei University Hospital, Malta, for providing us samples of Maltese breed.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
CONSORTIUM MEMBERS PA Marsan, V Balteanu, S Dunner, JF Garcia, C Ginja, J Kantanen
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Upadhyay, M., Chen, W., Lenstra, J. et al. Genetic origin, admixture and population history of aurochs (Bos primigenius) and primitive European cattle. Heredity 118, 169–176 (2017). https://doi.org/10.1038/hdy.2016.79
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2016.79
This article is cited by
-
Process and Dynamics of Mediterranean Neolithization (7000–5500 bc)
Journal of Archaeological Research (2022)
-
Extinct and Alive: Towards A Broader Account of Loss
Philosophia (2022)
-
On the origin and diversification of Podolian cattle breeds: testing scenarios of European colonization using genome-wide SNP data
Genetics Selection Evolution (2021)
-
Runs of homozygosity analysis of South African sheep breeds from various production systems investigated using OvineSNP50k data
BMC Genomics (2021)
-
Mutant alleles differentially shape fitness and other complex traits in cattle
Communications Biology (2021)
