
Abstract
Pancreas disease (PD), caused by a salmonid alphavirus (SAV), has a large negative economic and animal welfare impact on Atlantic salmon aquaculture. Evidence for genetic variation in host resistance to this disease has been reported, suggesting that selective breeding may potentially form an important component of disease control. The aim of this study was to explore the genetic architecture of resistance to PD, using survival data collected from two unrelated populations of Atlantic salmon; one challenged with SAV as fry in freshwater (POP 1) and one challenged with SAV as post-smolts in sea water (POP 2). Analyses of the binary survival data revealed a moderate-to-high heritability for host resistance to PD in both populations (fry POP 1 h2~0.5; post-smolt POP 2 h2~0.4). Subsets of both populations were genotyped for single nucleotide polymorphism markers, and six putative resistance quantitative trait loci (QTL) were identified. One of these QTL was mapped to the same location on chromosome 3 in both populations, reaching chromosome-wide significance in both the sire- and dam-based analyses in POP 1, and genome-wide significance in a combined analysis in POP 2. This independently verified QTL explains a significant proportion of host genetic variation in resistance to PD in both populations, suggesting a common underlying mechanism for genetic resistance across lifecycle stages. Markers associated with this QTL are being incorporated into selective breeding programs to improve PD resistance.
Similar content being viewed by others
Introduction
Infectious diseases present a significant threat to the sustainability of Atlantic salmon aquaculture (Wheatley et al., 1995; McLoughlin et al., 2002). Pancreas disease (PD), an alphaviral disease, is one of the most problematic infectious diseases, and causes high levels of mortality and morbidity on farms (FAO, 2013). Six subtypes of the PD-causing salmonid alphavirus (SAV) have been isolated in different parts of the world, including Scotland, Norway and Chile (Fringuelli et al., 2008). Subtypes are geographically specific, and farms within the same locality typically show infection with the same subtypes (Kristoffersen et al., 2009; Graham et al., 2012). For example, the two SAV subtypes in Norway (SAV2 and SAV3) have been shown to affect distinct sites (SAV2 in the north and SAV3 mainly in the south of Norway), with no overlap or co-infection within sites (Hjortaas et al., 2013; Jansen et al., 2014).
Natural infections with SAV have only been documented in the post-smolt stage of the salmon lifecycle, shortly after transfer from freshwater to sea water. Infection with SAV has been shown to result in histological changes in the heart, skeletal muscle and the pancreas of post-smolt salmon, as well as causing signs of morbidity such as a loss of appetite and lethargy (McLoughlin et al., 2002; Rodger and Mitchell, 2007; Taksdal et al., 2007). Response to infection may be influenced by many factors, such as feeding rate, season, temperature, stocking density, co-infection with other pathogens and host genetics (McLoughlin et al., 2002; Rodger and Mitchell, 2007; Graham et al., 2008; Norris et al., 2008; Kristoffersen et al., 2009; Jansen et al., 2010a; Stene et al., 2013). Long-term subclinical infections are common, and the peak in mortalities associated with a PD epidemic is often seen many months after infection (Karlsen et al., 2012). Survivors of infection can show chronic long-term illness and a reduced growth rate, leaving them vulnerable to infection with other pathogens and thus with drastically reduced economic value (Fringuelli et al., 2008).
Management techniques currently employed to prevent the spread of the virus and reduce morbidity and mortality levels include site fallowing, site hygiene measures and vaccination. A variety of vaccine types targeting the different SAV subtypes have been tested. However, these have shown a substantial variation in efficacy in test populations, suggesting a complex, multifactorial basis for protection (Rodger and Mitchell, 2007; Jansen et al., 2010a; Karlsen et al., 2012; Graham et al., 2014; Jansen et al., 2014). Consequently, there is a need for additional methods to complement or enhance current control measures, such as breeding salmon that are more resistant to PD.
Family-based selection for disease resistance, as a disease control strategy, has been an ongoing part of several Atlantic salmon breeding programs, with promising improvements in resistance seen for several diseases (Fjalestad et al., 1993; Storset et al., 2007). This strategy exploits the between-family genetic variation in resistance, where breeding candidates are selected based on performance of their siblings and other relatives in a disease challenge (Gjedrem, 1985; Gjøen and Bentsen, 1997). While effective, this approach ignores within-family variation, hence, will be less effective than approaches that utilise both within- and between-family genetic variation (Falconer and Mackay, 1996; Meuwissen and Goddard, 1996). For resistance (and other traits of economic value), the within-family variation can be exploited through the use of genetic markers as part of genomic or marker-assisted selection (GS and MAS respectively) in breeding programs. Both strategies require the identification of quantitative trait loci (QTL)-linked markers, which are used to identify the most resistant individuals within families (Meuwissen and Goddard, 1996). MAS is currently being implemented to increase host resistance to infectious pancreatic necrosis in Atlantic salmon breeding programs (Houston et al., 2008; Moen et al., 2009; Houston et al., 2010, 2012), and the potential of within-family GS using the large full-sibling families in aquaculture breeding programs is being explored (Sonesson and Meuwissen, 2009; Nirea et al., 2012).
Resistance to PD in Atlantic salmon has been shown to be moderately heritable (h2=0.21 (Norris et al., 2008)), hence, it is possible to apply family-based selection for enhanced PD resistance. However, as described above, improvements in selection accuracy, and therefore genetic gain, could be achieved through the use of genetic markers to capture within-family variation in resistance. Therefore, it is important to determine the genetic architecture of PD resistance and locate major QTL as candidates for MAS. Further, with reference genome sequences for salmonid species now available (Davidson et al., 2010; Berthelot et al., 2014), identification of genes underlying these major QTL may lead to an improved understanding of the biology of viral resistance in salmonid species.
The overall aim of this study was to explore the genetic architecture of PD resistance in Atlantic salmon, at both the fry and post-smolt stages. The specific aims of this study were to: (i) estimate the heritability of resistance to PD in large populations of Atlantic salmon fry and post-smolts challenged with SAV; (ii) detect and position QTL-influencing resistance to PD; and (iii) identify markers in population-level association with the resistance QTL, for potential use in MAS in these populations.
Materials and methods
Experimental populations
This manuscript describes two PD challenge studies carried out independently by two research groups in distinct populations of Atlantic salmon. One population was challenged as fry in freshwater and the other as post-smolts in sea water. As such, the population details, challenge protocol and analysis methodology have some specificities for each study. The first population (POP 1) consisted of 5558 Atlantic salmon fry from 218 full-sibling (FS) (83 paternal half-sibling; HS) families (~25 fry per FS family), from the 2010 year class of Marine Harvest. The Marine Harvest fish stock originates from the Mowi strain from the River Bolstad and the River Aaroy in Norway. Breeding programs using founder populations from these two rivers were established in the 1960 s (for further details, see Glover et al. (2009)). The second population (POP 2) consisted of 4946 post-smolts from 284 FS (120 paternal HS) families (~17 post-smolt per FS family) from the 2009 year class of SalmoBreed AS. The founding populations of the SalmoBreed fish stock originate from the Bolaks and Jakta strains, and breeding programs were established in the 1970 s (see http://www.salmobreed.no/en/about/company-history). Therefore, the two challenge populations have distinct genetic backgrounds.
SAV challenges
Both populations (POP 1 and POP 2) were challenged with the same strain of SAV, SAV3, which is the most abundant strain in Norway (Hodneland et al., 2005). However, due to the different lifecycle stages of the populations, the challenge protocols were different. Both challenge experiments were carried out in accordance with guidelines from the Norwegian Animal Research Authority by VESO Vikan, Norway (POP 1) and PHARMAQ AS, Norway (POP 2).
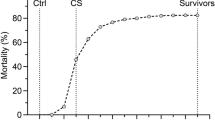
In the POP 1 challenge, fry at 51 days post hatch (average weight: 0.5 g) were starved for 24 h prior to challenge. The challenge protocol used was as follows: 100 Atlantic salmon parr (‘shedders’; average weight ~38 g) were infected with SAV3 using an intraperitoneal injection. The viral challenge dose was 3.3e5 TCID50 per shedder. Infected parr were kept in a tank of 216 l in volume and were allowed to shed virus into this tank for one week. Effluent water from this tank was passed into a single fry challenge tank of 10 l in volume. Water temperature in this fry challenge tank was maintained at 12 °C and water flow was >1 l Kg−1min−1 to ensure >75% O2 saturation in water effluent. The challenge commenced on 15 June 2010 and continued until mortalities were negligible (end date: 11 August 2010). Ten fry from the main mortality period (that is, after >10% mortality was obtained) were sampled and measured for viral load (quantitative PCR). The total mortality level at the end of the challenge was 3456 (60%) (Figure 1a). Mortalities were removed from the tank daily, whereas survivors were collected and euthanized at the end of the experiment (in accordance with guidelines from the Norwegian Animal Research Authority). Tissue samples from both mortalities and survivors were stored at −20 °C. For all further analyses conducted in POP 1 (including parentage assignment), PD mortalities were defined as those which occurred between dates 5 July 2010 (21 days post challenge) and 04 August 2010 (8 days prior to challenge termination) (Figure 1a).

Mortality profiles. (a) POP 1 (fry)—number of mortalities observed per day over the course of the challenge, from 18 days post challenge (2 June 2010—prior to which mortalities were negligible) to the challenge termination date (11 August 2010). The peak in mortalities was observed 28 days post challenge. For all analyses in POP 1, mortalities between dates 5 June 2010 (21 days post challenge) and 4 August 2010 (51 days post challenge) were assumed to be due to PD, and the mortality profile within this time frame is indicated by the dotted red lines. (b) POP 2 (post-smolts)—number of mortalities observed per day over the challenge duration. The peak in mortalities occurred on day 13 post challenge. Further analyses in POP 2 were conducted using mortalities which occurred 7 days post infection; these mortalities are highlighted by the dotted red lines. (c) POP 1 (fry)—the number of full-sibling families across the 150 full-sibling families with a given percentage mortality. Family mortality ranged from 0 to 100%, with an average mortality of 61%. These families were used to estimate the heritability of resistance to PD. (d) POP 2 (post-smolts)—total number of full-sibling families with a given percent mortality. Family mortality ranged from 5 to 100%, with an average family mortality of 62%.
In the POP 2 challenge, post-smolts at body weight ~85 g and 333 days post hatch were infected with SAV3 by means of direct intraperitoneal injection. Each fish was injected with 0.1 ml of the SAV3 virus isolate provided by PHARMAQ AS (see Karlsen et al. (2012) for further details). Injected test fish were kept in saltwater in a single 4 m diameter tank for the duration of the challenge. The water temperature was maintained at 12 °C. Water flow was >1.7 l kg−1min−1 and was continuously adjusted to ensure >80% oxygen saturation in water effluent. The challenge commenced on 7 December 2009 and continued until mortalities were negligible (end date: 22 December 2009). The total mortality at the end of the challenge was 3058 (62%) (Figure 1b). Mortalities were recorded and sampled once daily, and survivors were collected and euthanized at the end of the experiment (in accordance with guidelines from the Norwegian Animal Research Authority). Ventricle samples from 20 mortalities from day 9 to day 14 post challenge were sent for real-time PCR to detect the presence of SAV and quantify viral load. Tissue samples from each fish were taken and stored at −20 °C for subsequent DNA extraction and genotyping. For all further analyses, PD mortalities were defined as those occurring between 7 days post challenge, where the mortality level rose substantially above baseline levels, to the end of the epidemic (16 days post challenge), when mortalities were negligible.
Parentage assignment
As the challenged fry in POP 1 were too small to be tagged (for example, using an electronic passive integrated transponder (PIT) tag), parentage assignment of surviving and mortality offspring was undertaken by genotyping. Survivors were genotyped using a panel of nine microsatellite loci and assigned to family (GenoMar, Co/Glastad Invest AS, Fridtjof Nansens Plass 3, Oslo, Norway). Parentage assignment of mortalities was carried out at The Roslin Institute using a panel of 87 single-nucleotide polymorphisms (SNPs) distributed across all chromosomes (taken from Moen et al. (2008)). Eighteen of these failed the genotyping assay or were uninformative, thus 69 SNPs remained for further analyses (Supplementary Table 1) (genotyping was performed by LGC Genomics Ltd, Herts, EN11 0WZ, UK). Three software packages were used for parentage assignment of mortalities: SNPPIT (maximum likelihood algorithm) (Anderson, 2010), FAP (genotype exclusion algorithm) (Taggart, 2007) and Vitassign (genotype exclusion algorithm) (Vandeputte et al., 2006). Successful parentage assignment of mortalities was taken only if agreement was seen between outputs from at least two of the software.
In POP 2, all fish were individually PIT tagged prior to the mixing of families (at ~19 g body weight) and, therefore, PIT tag scanning of surviving and mortality fish was used for family assignment.
Quantitative genetic parameter estimation
POP 1–PD resistance of fry
One hundred and fifty FS families (72 HS families; 72 sires and 150 dams; parents were not closely related) with >15 offspring (total of survivors and mortalities; average mortality per family: 61%; Figure 1c) were selected for estimation of genetic parameters, that is, the additive genetic variation and heritability for PD resistance. Variance components were estimated by fitting the following linear mixed model in the ASReml software (Gilmour et al., 2009):

where Yijk is the observed SAV challenge outcome for individual k with sire i and dam j; μ is the population mean; Si and Dij are the random additive genetic effects of the ith sire and jth dam; and eijk is the residual variance. Sire and Dam were fitted as random effects and assumed to be normally distributed, with variances σ2S and σ2D, respectively. The total additive genetic variance was estimated as 2(σ2S+σ2D). Common environmental effects associated with FS family were not significant, hence, this effect was not fitted.
As challenge outcome was scored as a binary variable (survived or died), the heritability of PD resistance was estimated on the observed binary scale, and also by using a logit-link or probit-link function to account for the binary data. Assuming a continuous underlying liability for the binary challenge outcome, the observed binary scale heritability (h201) was converted to the underlying liability scale using the formula (from Falconer and Mackay (1996)):

where p is the proportion of dead individuals in the dataset, and i is the mean deviation (in s.d. units) from the population mean of the highest ranking p individuals, assuming a normal distribution.
POP 2–PD resistance of post-smolts
Estimation of genetic parameters in POP 2 was conducted using all 284 FS families described in the challenge protocol above. Twenty-nine mortalities which occurred prior to day 7 of the challenge were excluded as they occurred before the PD outbreak (Figure 1b). As in POP 1, the trait analysed was binary PD challenge outcome (survived or died) at the challenge termination date. Two models were applied to estimate variance components using the ASReml software (Gilmour et al., 2009). Model 1 was an animal linear model:

where Yij is the observed survival status in the challenge test of individual i from FS family j; μ is the population mean; twi is the body weight of individual i at tagging; β is a first degree fixed regression coefficient of survival on twi; ai is the random additive genetic effect of individual i; cj is the random effect common to FS family j other than additive genetics (that is, environmental effect of common rearing tank prior to PIT tagging, non-additive genetic effects, and maternal effects); and eij is the individual random residual effect.
Model 2 was a linear sire-dam model:

where Yijk is the observed survival status in the challenge test of individual i in FS family jk with sire j and dam k; and Sj and Djk are the random effects of the jth sire and kth dam. As sires and dams were selected from the same population, the sire and dam additive genetic variances (σ2S and σ2D, respectively) were constrained to be equal (σ2S=σ2D=¼σ2a). From model 1, the heritability was estimated as follows:

whereas in model 2 it was estimated as:

In both cases, the non-additive effect common to FS was estimated as:

where σ2a is the additive genetic variance, σ2S is the additive genetic sire variance, σ2c is the non-additive variance common to FS; σ2e is the residual variance; and σ2p=σ2a+σ2c+σ2e for results from model 1 and σ2p=2σ2S+σ2c+σ2e for results from model 2. As in POP 1, heritabilities on the observed binary scale obtained in both model 1 and model 2 were converted on to the underlying liability scale, to account for the binary format of challenge outcome. In addition to this, model 2 was fitted on the probit scale, using the probit-link function in ASReml.
Genotyping for QTL mapping
For POP 1, a two-step QTL mapping strategy was employed to utilise the disparity in recombination rate between the sexes in Atlantic salmon (Hayes et al. (2006); Houston et al. (2008); described in more detail below). QTL detection was performed using a sparse SNP panel and sire-based mapping, whilst QTL confirmation and estimation of position was performed using a denser SNP panel and dam-based mapping. From the 150 FS families used for estimation of genetic parameters, 20 paternal HS families (55 FS families) with intermediate levels of mortality (range: 45–72%) were selected for QTL mapping. Survivors from these families were identified, their genomic DNA was extracted (using the Qiagen DNeasy 96 protocol Blood & Tissue kit), and genotyped (by LGC Genomics Ltd) for the same sparse SNP panel used for parentage assignment of mortalities (Supplementary Table 2). One hundred and seventy-seven survivors (28%) with poor quality genotyping information (excess missing genotypes or Mendelian errors given expected family assignment conducted using the microsatellite panel) were removed from the QTL mapping analysis.
In POP 2, genomic DNA was extracted from fin and muscle samples of 1182 parents and offspring, using a modified high-salt precipitation protocol (http://www.genomics.liv.ac.uk/animal/RESEARCH/ISOLATIO.PDF). DNA was quantified using the PicoGreen double-strand DNA quantitation assay (Invitrogen, Life Technologies LTD, Paisley, UK), and DNA concentrations normalised to 50 ng μl−1. Genotyping was carried out according to the manufacturer's instructions, using the 6 K Atlantic salmon Illumina iSelect SNP array developed by the Centre for Integrative Genetics (www.cigene.no). Genotype data were quality-checked using Genome Studio (Illumina Inc, San Diego, CA, USA) and were filtered to achieve a 95% call rate for both samples and SNPs. SNPs were filtered to retain those with MAF >2.5%. The remaining 5278 SNPs were further filtered to remove those with low quality and those with a MAF of <1%, leaving 4178 SNPs for subsequent analyses.
QTL mapping
For both populations, QTL mapping was conducted in the GridQTL software (Allen et al., 2012) using two methods. First, taking advantage of the large paternal HS families, a HS analysis was conducted in both populations independently, which uses a linear regression-based interval mapping approach to QTL identification (for details, see Knott et al. (1996)). In brief, using a multiple marker-based approach, the probability of inheriting a particular allele at a particular marker location is calculated and used to estimate the information content for each marker. At each centiMorgan (cM) interval, the phenotypes (binary challenge outcome) are regressed on the probabilities of inheriting particular alleles. The strength of evidence for a QTL is calculated, expressed as an F ratio, with the numerator degrees of freedom equal to the number of parents that are informative at a given chromosome (Knott et al., 1996). To determine whether the estimated QTL F ratio was significant (that is, whether the identified QTL was significantly influencing resistance) at the chromosome- and/or genome-wide level, chromosome- and genome-wide critical F ratios were calculated using permutation testing in the GridQTL software, as follows. The chromosome-wide critical F ratio threshold was determined using 10 000 permutations in both populations. A genome-wide critical F ratio threshold was calculated by first obtaining a Bonferroni corrected P-value at the 5% significance level (given the 29 pairs of chromosomes of Atlantic salmon, adjusted P-value=0.05/29) and then obtaining the genome-wide F ratio threshold at this adjusted P-value, using 10 000 permutations (Churchill and Doerge, 1994). The QTL F ratio was compared with the chromosome- and genome-wide critical F ratio, and if the QTL F ratio was larger than either one of these, then this QTL was determined as significantly affecting PD resistance in this population. For each genome-wide significant QTL, confidence intervals for the location parameter were estimated using the bootstraps with resampling method and 10 000 iterations (Visscher et al., 1996).
Second, to exploit the FS data set structure in both populations (family sizes of 15–29 offspring in POP 1 and 4–37 offspring in POP 2), QTL mapping was also conducted in both populations independently using a sib-pair (SP) approach, implemented in the GridQTL software. This approach is based on the principle that FS who inherit more QTL alleles identical-by-descent (IBD) tend to be more similar in phenotype, that is, the difference between their phenotypes tends to be smaller the more QTL alleles they share IBD (Haseman and Elston, 1972). IBD probabilities are first calculated at 1 cM intervals, and the residuals from this analysis are then used in a Haseman–Elston approach, in which the squared difference of the phenotypes (that is, residuals) is regressed on to the IBD probabilities (Haseman and Elston, 1972; Knott and Haley, 1998). QTL significance and confidence intervals in both populations was determined using permutation testing, as described for the HS analysis above.
The proportions of within-family variance explained (PVE) by each significant QTL were estimated using the HS analyses in GridQTL, and the following formulae. For the sire- or dam-based linkage analysis: h2QTL=4[1-(MSEfull/MSEred)]; for the combined sire- and dam-based linkage analysis: h2QTL=2[[1-(MSEfull/MSEred)SIRE]+[1-(MSEfull/MSEred)DAM]]; where MSEfull and MSEred are the mean square errors for the models with and without the QTL, respectively.
Despite the similarity of the statistical methods used to identify QTL, different genotyping approaches were implemented in the two different populations. Therefore, some details of the QTL-mapping analyses were population-specific, and these are described below.
The HS QTL-mapping analysis in POP 1 was carried out using a two-step approach (Hayes et al., 2006; Houston et al., 2008), which takes advantage of the large disparity in recombination rates between Atlantic salmon males and females. The low recombination rate in males means that the inheritance of large sections of the genome from sire to offspring can be tracked by genotyping individuals using a sparse marker set. This allows the identification of chromosomes harbouring QTL significantly influencing the trait of interest (step one). Significant QTL can then be positioned on chromosomes by genotyping a denser set of markers only for the significant chromosomes, and following the inheritance patterns of these markers from dams to offspring (step two).
This two-step approach for QTL identification was applied in POP 1, using 1273 offspring (463 survivors, 810 mortalities) belonging to 20 paternal HS (55 FS) families (HS family size range: 41–90 offspring; average=64) with intermediate levels of mortality (mortality range 40–70%). Assuming QTL of intermediate frequency, the use of paternal HS families with intermediate mortality levels should, in principle, increase the power for the detection of QTL segregating within families. A dam-linkage analysis using the same approach and sparse SNP panel (55 dams, resulting in 55 FS families, with average size 23 offspring per family) was also conducted to identify QTL segregating in dams but not sires, recognising that dam-segregating QTL may be missed with sparse markers. In the second step of QTL mapping, a denser set of SNP markers (36 in total, taken from Lien et al. (2011) and Gonen et al. (2014) and including the sparse marker panel used in step one; Supplementary Table 2) was genotyped for chromosomes identified as containing QTL at chromosome- or genome-wide significance in step one. Linkage maps for these chromosomes were constructed using the Lep-MAP software (Rastas et al., 2013) (Supplementary Table 2). Using this denser set of mapped SNPs, a dam-based linkage analysis was conducted to test for the presence of QTL and to estimate their position on the chromosome. The SP analysis in POP 1 was first conducted for all 29 chromosomes using the sparse set of SNP markers. For chromosomes with the denser set of markers genotyped (that is, were significant in the sire-based HS analysis), the SP analysis was repeated using the denser set of SNPs.
In POP 2, male and female mapping parents were used in a combined ‘HS’ analysis. To convert the genotype data sets into a format compatible with the GridQTL mapping software, each data set was duplicated prior to analysis, with the designation of parents as sire or dam inverted in the duplicate. All analyses were carried out using the genetic map produced by Lien et al. (2011), with the exception of chromosome 3; the chromosome for which a genome-wide significant QTL was identified. For this chromosome, a separate map was produced based on the data generated within this study using CRIMAP (version 2.4 (Green et al., 1990), modified by Xuelu Liu (Monsanto)), and the results for this chromosome are referred to relative to this map.
Association analysis
A genome-wide association analysis between SNP markers and PD mortality was performed in POP 2 (where the 6K Atlantic salmon SNP array was used for genotyping). The quality-filtered 4178 SNPs (retaining those of high quality and MAF>1%) were used to conduct an association analysis in the GenABEL software package (Aulchenko et al., 2007). Following the recommended procedure from the software manual, inflation caused by stratification was calculated, and when the inflation measurement was different from 1, the pedigree kinship matrix was calculated and fitted to the data, using the function ‘polygenic()’. The residuals from the 'polygenic()' function were then used as input (without covariates) to the 'qtscore()' function for the association analysis. The ‘qtscore()’ function implements the association test ‘Genome-wide Rapid Analysis using Mixed Models And Score’ (GRAMMAS, Amin et al. (2007)). Test significance was reported as both the P-values calculated by the package individually for each SNP, and also adjusted P-values based on permutation tests to empirically estimate the genome-wide significance. In POP 1, individual SNPs on the QTL-containing chromosomes were also tested for association with PD resistance, using the software package ASReml (Gilmour et al., 2009). The SNP association was conducted by fitting the same mixed model used to estimate the heritability, with the added step of fitting each SNP individually as a fixed effect.
Results
Challenge outcomes and parentage assignment
For all analyses conducted in POP 1, mortalities between 21 and 51 days post challenge were assumed to be due to PD, as described above. Overall, 3456 mortalities were observed, 3415 of which were designated as PD mortalities. Of these, 2455 were successfully assigned to 71 HS (157 FS) families. 2328 survivors remained at the challenge termination date (11 August 2010) (Figure 1a), 2102 of which were successfully assigned back to 81 HS (203 FS) families.
In the post-smolt challenge (POP 2), mortalities occurred over a much shorter period, hence the trial duration was shorter (Figure 1b). Overall, 3058 mortalities and 1888 survivors were observed across the 16-day challenge (7 December 2009 to 22 December 2009). All survivors and 3029 mortalities occurring 7 days post infection (14 December 2009 to 22 December 2009) were included in subsequent analyses. Real-time PCR results of the 20 sampled mortalities were all positive for PD virus (Ct range 15.0–19.5). Given that each fish had been individually PIT tagged prior to challenge, parentage assignment by genotyping was not required.
Estimated heritabilities
The heritability for PD resistance in POP 1 was estimated using 72 HS (150 FS) families with 15 or more offspring (3949 offspring in total, 2367 mortalities, 1582 survivors). Mortalities in these families ranged from 0 to 100%, with an average mortality of 61% (Figure 1c). The heritability of PD resistance across these families was estimated at 0.34 on the observed binary scale, which equated to ~0.5 on the underlying liability scale (Table 1), and this estimate was relatively consistent across the different models (underlying liability, logit-link and probit-link models). Non-genetic effects associated with family were not significant.
In POP 2, heritability estimates were obtained from a data set consisting of 4917 individuals belonging to 284 FS families. The average mortality per FS family at the challenge termination date was 62% (Figure 1b), and mortalities per family ranged from 5 to 100% (Figure 1d). Using these families, heritabilities of 0.26 and 0.23 were estimated on the observed binary scale using two models: Model 1—Animal model; Model 2—Sire-dam model (Table 1). As with POP 1, these heritabilities were transformed on to the underlying liability scale, and a heritability of ~0.4 was obtained using both models (Table 1). Fitting model 2 on the probit-link scale resulted in a heritability estimate of 0.33, which is consistent with the underlying liability scale heritability estimate of 0.37 (Table 1). Estimated effects common to FS other than additive genetics were small (range: 0.03–0.05; Table 1) and not significant.
QTL mapping
In POP 1, QTL were identified by applying a two-step mapping approach in the HS analysis, using a sparse then a denser SNP panel, and further taking advantage of the unequal recombination rates between males and females (described in Materials and Methods). The initial sire-linkage analysis using a sparse SNP panel identified three putative QTL affecting PD resistance, on chromosomes 3, 7 and 23 (Table 2). The QTL on chromosome 23 was significant at the genome-wide level, whereas the QTL on chromosomes 3 and 7 were significant at the chromosome-wide level. The QTL on chromosome 3 was confirmed with a dam-linkage analysis using the sparse SNP panel, which also identified a further QTL on chromosome 4 (both were significant at the chromosome-wide level). The sib-pair (SP) analysis using the same sparse SNP panel also identified the QTL on chromosomes 3 and 4, with the QTL on chromosome 4 reaching genome-wide significance (Table 2).
To estimate the position of QTL on the chromosomes, a further 28 SNPs were genotyped across the four chromosomes for which QTL were identified at the chromosome- or genome-wide significance level (Table 2), and these were then used in a dam-linkage HS analysis. This confirmed and positioned QTL on chromosomes 3 and 4 towards the ends of the linkage maps, at map positions 135 cM and 74 cM, respectively (Table 2; chromosome 3 position shown in Figure 2a). The SP analysis using the denser set of markers confirmed the QTL on chromosomes 3 and 4 (both reached chromosome-wide significance), and estimated QTL locations overlapped with those obtained from the HS analysis (chromosome 3 at 129 cM and chromosome 4 at 75 cM). The confidence interval for the QTL on chromosome 4 was narrowed to 13 cM in the SP analysis, using bootstrapping.

Chromosome 3 QTL location. The estimated position of the QTL on chromosome 3 in all analyses. The green dotted lines represent chromosome-wide F ratio thresholds, and the red dotted lines represent genome-wide F ratio thresholds. Panels a and b show the position of the QTL on chromosome 3 in POP 1, identified using the dense SNP marker panel in the HS and SP analyses, respectively. Panels c and d are the positions of the QTL on chromosome 3 for POP 2, for HS and SP analyses, respectively. The overlap in QTL position is shown in panel e, with map positions given according to the POP 2 map.
Overall, the four QTL were individually estimated to explain between 4 and 9% of the within-family variance for PD resistance (Table 2). The proportion of within family variance estimated using the sire-based linkage analysis only were comparable between the three sire-segregating QTL. The QTL on chromosome 3 was the only QTL identified in both the sire- and dam-based linkage analysis, and explained the highest proportion of within-family variance for resistance when both sire- and dam-based analyses were considered together.
In POP 2, one genome-wide significant QTL was detected in both the HS and SP analyses, localised at the distal end of chromosome 3 based on the map by Lien et al. (2011) (Table 2 and Figures 2b and c). The QTL peaks in the SP and HS analyses were concordant (QTL peaks at 61 cM and 72 cM, respectively; Table 2). The apparent 10 cM difference in QTL peak in the two analyses is likely to be a result of the SP analysis not allowing identical map locations for adjacent markers. In these cases, a small arbitrary genetic distance was inserted, which resulted in an expansion of the overall map. In the HS analysis, the QTL on chromosome 3 explained ~23% of the within-family variance for survival, and was found to be segregating in 18 of 99 parents. The most likely position of the QTL was towards the distal end of the chromosome (Figure 2c). In addition to the genome-wide significant QTL on chromosome 3, two suggestive, chromosome-wide significant QTL were detected, on chromosome 2 (HS and SP) and chromosome 14 (SP) (Table 2).
Across both populations and all analyses, chromosome 3 was identified as containing a resistance QTL. To confirm that this QTL maps to the same region of chromosome 3 in both populations, common markers between the linkage maps used in QTL mapping in the two populations were identified, and used as anchors to orientate the maps relative to each other. Likelihood profile maps were plotted to demonstrate that the QTL mapped to the same region of chromosome 3 (Figure 2e). This suggests a common QTL-influencing resistance in both populations, and across both the fry and post-smolt stages of the Atlantic salmon lifecycle.
Association analysis
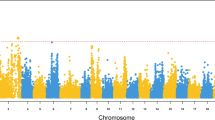
In POP 2, all included post-smolts were genotyped using the 6 K Atlantic salmon SNP array. To detect SNPs showing significant association to PD resistance in POP 2, a genome-wide association analysis was performed, using 4178 quality-filtered SNPs. Of these, 184 SNPs reached nominal significance P<0.05, of which 20% were found on chromosome 3 (Figure 3). After permutation testing to estimate genome-wide significance levels, two SNPs were significant (adjusted P<0.01). Both SNPs are located on chromosome 3, the most significant at 120 cM (ESTV_18493_1052) and the second at 113 cM (ESTNV_29410_233), using the map generated for this chromosome by the data in this study. In the published linkage map, the female/male positions for these two SNPs are 111/58 cM and 104/57 cM, respectively (Lien et al., 2011). In POP 1, two SNPs on chromosome 3 were also significantly associated (P<0.001) with resistance to PD: consensus46559_59 and consensus110127_55 at map positions 67 and 87 cM, respectively.

Genome-wide association analysis in POP 2. Manhattan plot depicting P-values (corrected for inflation) from the 1-d.f. test for association between SNP and trait (binary survival) for (a) the whole genome (29 chromosomes) and (b) chromosome 3.
Discussion
To explore the genetic architecture of PD resistance, two independent populations of farmed Atlantic salmon fry and post-smolts were challenged using SAV3, the most prevalent strain of SAV in Norway. Using the binary trait of mortality/survival from the challenge experiments, moderate-to-high heritability estimates of resistance to PD were obtained (~0.5 for fry in POP 1 and ~0.4 for post-smolt in POP 2). Following this, QTL mapping studies in the two populations identified six putative resistance QTL. The most convincing and robust evidence for a QTL in both populations was detected near the distal end of chromosome 3. This QTL explained the largest proportion of within-family variance for PD resistance in both populations, and markers showing population-level association with PD resistance were identified close to the QTL peak.
The high heritability obtained from fry in POP 1 in this study is similar to some of the larger estimates reported for disease resistance traits in Atlantic salmon (for example, 0.55 for infectious pancreatic necrosis and 0.51–0.62 for furunculosis (Kjøglum et al., 2008; Drangsholt et al., 2011)). Notably, this estimate is almost double than that obtained by Norris et al. (2008) (h2=0.21), where a natural SAV outbreak in farmed Atlantic salmon smolts was analysed. The heritability estimate obtained in post-smolts for POP 2 in the current study is moderate-to-high (0.33 from the probit analysis, ~0.4 when transformed from linear model analysis), but still larger than the estimate obtained by Norris et al. (2008). Despite the differences (including life stage, challenge model and population origins) between the three populations (POP 1 and POP 2 in this study, and the population in Norris et al. (2008)), there is consistent evidence for high heritable variation in PD resistance. This, combined with the large variation in mortality seen in both populations in this study (POP 1 and POP 2), strongly suggests that selection for PD resistance is plausible.
In both POP 1 and POP 2, subsets of families showing intermediate levels of mortality were deliberately chosen for QTL analysis from a larger set of families. This increases the power of QTL detection by linkage analysis by increasing the likelihood of having QTL-segregating parents in the data set, and reduces the false-positive rate in genome-wide association analysis by minimising spurious associations resulting from phenotypically extreme families (Darvasi and Soller, 1992; Hayes et al., 2009). The two studies did, however, differ in the marker genotyping strategy and hence also in the QTL mapping approaches, as described above. In simple terms, in POP 1 the experiment was designed to exploit the sex-specific recombination rates to reduce the number of individual SNPs required (and therefore increase efficiency), whereas in POP 2 this was unnecessary because all animals were genotyped for a SNP chip (Lien et al., 2011). However, despite the differences in genotyping technologies and methods of analysis within (HS vs SP) and between the populations, one single QTL on chromosome 3 was repeatedly identified as chromosome- or genome-wide significant in all analyses. This not only provides validation and strong support for this QTL, but also highlights the efficacy of both genotyping and mapping approaches.
Natural outbreaks of PD are observed almost exclusively at the post-smolt stage of the Atlantic salmon lifecycle. Although viral isolates have been detected in freshwater, no outbreaks of PD in the fry stage have been recorded (Jansen et al., 2010b). The practicality of large-scale challenge experiments at the fry stage of the salmon lifecycle has meant that fry challenges can be used as a model for post-smolt PD outbreaks (Cano et al., 2014). However, differences in behaviour, environment and physiology between the freshwater and marine stages of the salmon lifecycle mean that the genetic architecture of PD resistance could potentially differ between the two life stages.
The identification of the same QTL on chromosome 3 in both the fry and post-smolt populations in this study suggests that similar biological mechanisms are likely to be influencing this component of resistance to PD in both lifecycle stages. If the causal factors underlying the QTL on chromosome 3 are related to an immune response, they are likely to be part of a general innate immune response against SAV, as the adaptive immune response is undeveloped at the juvenile stage. A significant induction of innate genes, such as IFN and Mx, has been implicated in the response of salmon to SAV challenge (Grove et al., 2013; Herath et al., 2013). Alternatively, the causal factor(s) underlying the resistance QTL may affect variation in the ability of the host to block the progression of the viral lifecycle. The concordance of the QTL between the two populations (one intraperitoneally injected and the other given a form of bath challenge) suggests that the main underlying factors are unlikely to be related to barrier function. However, the lack of concordance between the other QTL identified in the two studies suggests that there may also be factors underlying genetic resistance that are unique to each lifecycle stage and challenge model.
MAS has been successfully applied in aquaculture breeding, including for female monosex Chinook salmon production (Devlin et al., 1991), for resistance to lymphocystis disease in Japanese flounder (Fuji et al., 2007), and for resistance to infectious pancreatic necrosis in Atlantic salmon (Houston et al., 2008; Moen et al., 2009; Houston et al., 2010). In this study, SNPs in putative linkage disequilibrium with the PD resistance QTL on chromosome 3 have been identified, and are currently being employed in MAS to select for improved PD resistance in commercial Atlantic salmon (H Bakke, pers. comm.). However, it is possible that these markers are a considerable distance from the causative gene and/or mutation. Further refinement of the QTL position and eventual identification of candidate genes or mutations would be advantageous for both applied MAS (more accurate marker predictors of QTL genotype) and our understanding of the biological basis of genetic resistance to PD. This could be achieved by genotyping and testing a much higher number of SNPs in the region of the QTL, for example using a high-density SNP array (Houston et al., 2014), or by re-sequencing of alternate homozygotes at the QTL on chromosome 3. In addition, positional and functional candidate genes for the QTL can be generated by taking a comparative genomics approach, as demonstrated for Infectious Salmon Anemia resistance (Li et al., 2011), or by interrogation of the Atlantic salmon genome (Davidson et al., 2010) as its assembly and annotation improves.
Conclusions
Two independent populations of Atlantic salmon were challenged with SAV3; one as fry in freshwater and the other as post-smolts in marine water. A moderate-to-high heritability for resistance to PD was estimated in both populations (fry h2~0.5; post-smolt h2~0.4), demonstrating the feasibility of family selection for PD resistance. QTL mapping analyses identified six chromosomes (2, 3, 4, 7, 14 and 23) harbouring putative PD resistance QTL across both populations. A QTL on the distal end of chromosome 3 was identified in all analyses and explained the largest proportion of within-family variation in resistance in both populations. The detection of this QTL across both the fry and post-smolt stages suggests a common mechanism for PD resistance at both lifecycle stages. SNPs on this chromosome were significantly associated with PD resistance at a population level, and these are currently being implemented in MAS for improved PD resistance. Further refinement of this result may lead to more effective marker-based selection and an improved understanding of the host regulation of resistance to this important disease.
Data Archiving
Data available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.5241h.
References
Allen J, Scott D, Illingworth M, Dobrzelecki B, Virdee D, Thorn S et al. (2012) CloudQTL: Evolving a Bioinformatics Application to the Cloud. Digital Research 2012, 10–12 September 2012: Oxford, UK.
Amin N, van Duijn CM, Aulchenko YS . (2007). A genomic background based method for association analysis in related individuals. PLoS One 2: e1274.
Anderson EC . (2010). Computational algorithms and user-friendly software for parentage-based tagging of Pacific salmonids. Final report submitted to the Pacific Salmon Commission's Chinook Technical Committee (US Section) 46 pages.
Aulchenko YS, Ripke S, Isaacs A, Van Duijn CM . (2007). GenABEL: an R library for genome-wide association analysis. Bioinformatics 23: 1294–1296.
Berthelot C, Brunet F, Chalopin D, Juanchich A, Bernard M, Noël B et al. (2014). The rainbow trout genome provides novel insights into evolution after whole-genome duplication in vertebrates. Nat Commun 5: 3657.
Cano I, Joiner C, Bayley A, Rimmer G, Bateman K, Feist SW et al. (2014). An experimental means of transmitting pancreas disease in Atlantic salmon Salmo salar L. fry in freshwater. J Fish Dis 38: 271–281.
Churchill GA, Doerge RW . (1994). Empirical threshold values for quantitative trait mapping. Genetics 138: 963–971.
Darvasi A, Soller M . (1992). Selective genotyping for determination of linkage between a marker locus and a quantitative trait locus. Theor Appl Genet 85: 353–359.
Davidson WS, Koop BF, Jones SJM, Iturra P, Vidal R, Maass A et al. (2010). Sequencing the genome of the Atlantic salmon (Salmo salar. Genome Biol 11: 403.
Devlin RH, McNeil BK, Groves TDD, Donaldson EM . (1991). Isolation of a Y-chromosomal DNA probe capable of determining genetic sex in Chinook salmon (Oncorhynchus tshawytscha. Can J Fish Aquat Sci 48: 1606–1612.
Drangsholt TMK, Gjerde B, Odegard J, Finne-Fridell F, Evensen O, Bentsen HB . (2011). Quantitative genetics of disease resistance in vaccinated and unvaccinated Atlantic salmon (Salmo salar L.). Heredity 107: 471–477.
Falconer DS, Mackay TFC . (1996) Introduction to Quantitative Genetics, 4th edn, London, UK: Longmann & Co.
FAO. (2013). Cultured Aquatic Species Information Programme. Salmo salar (Linnaeus, 1758). [online] Available at <http://www.fao.org/fishery/culturedspecies/Salmo_salar/en#tcNA00D6< (accessed 26 September 2013).
Fjalestad KT, Gjedrem T, Gjerde B . (1993). Genetic improvement of disease resistance in fish: an overview. Aquaculture 111: 65–74.
Fringuelli E, Rowley HM, Wilson JC, Hunter R, Rodger H, Graham DA . (2008). Phylogenetic analyses and molecular epidemiology of European salmonid alphaviruses (SAV) based on partial E2 and nsP3 gene nucleotide sequences. J Fish Dis 31: 811–823.
Fuji K, Hasegawa O, Honda K, Kumasaka K, Sakamoto T, Okamoto N . (2007). Marker-assisted breeding of a lymphocystis disease-resistant Japanese flounder (Paralichthys olivaceus. Aquaculture 272: 291–295.
Gilmour AR, Gogel BJ, Cullis BR, Thompson R . (2009) ASReml User Guide Release 3.0. VSN International Ltd, Hemel Hempstead, HP1 1ES: UK.
Gjedrem T . (1985). Improvement of productivity through breeding schemes. GeoJournal 10: 233–241.
Gjøen HM, Bentsen HB . (1997). Past, present, and future of genetic improvement in salmon aquaculture. ICES J Mar Sci 54: 1009–1014.
Glover KA, Otterå H, Olsen RE, Slinde E, Taranger GL, Skaala Ø . (2009). A comparison of farmed, wild and hybrid Atlantic salmon (Salmo salar L.) reared under farming conditions. Aquaculture 286: 203–210.
Gonen S, Lowe NR, Cezard T, Gharbi K, Bishop SC, Houston RD . (2014). Linkage maps of the Atlantic salmon (Salmo salar genome derived from RAD sequencing. BMC Genomics 15: 166.
Graham DA, Wilson C, Jewhurst H, Rowley H . (2008). Cultural characteristics of salmonid alphaviruses - influence of cell line and temperature. J Fish Dis 31: 859–868.
Graham DA, Fringuelli E, Rowley HM, Cockerill D, Cox DI, Turnbull T et al. (2012). Geographical distribution of salmonid alphavirus subtypes in marine farmed Atlantic salmon, Salmo salar L., in Scotland and Ireland. J Fish Dis 35: 755–765.
Graham DA, Rowley HR, Frost P . (2014). Cross-neutralization studies with salmonid alphavirus subtype 1-6 strains: results with sera from experimental studies and natural infections. J Fish Dis 37: 683–691.
Green P, Falls K, Crooks S . (1990) Documentation for CRI-MAP, version 2.4. Washington School of Medicine: St Louis, MO, USA.
Grove S, Austbo L, Hodneland K, Frost P, Lovoll M, McLoughlin M et al. (2013). Immune parameters correlating with reduced susceptibility to pancreas disease in experimentally challenged Atlantic salmon (Salmo salar. Fish Shellfish Immunol 34: 789–798.
Haseman JK, Elston RC . (1972). Investigation of linkage between a quantitative trait and a marker locus. Behav Genet 2: 3–19.
Hayes BJ, Gjuvsland A, Omholt S . (2006). Power of QTL mapping experiments in commercial Atlantic salmon populations, exploiting linkage and linkage disequilibrium and effect of limited recombination in males. Heredity 97: 19–26.
Hayes BJ, Macleod IM, Baranski M . (2009). Sampling strategies for whole genome association studies in aquaculture and outcrossing plant species. Genet Res 91: 367–371.
Herath TK, Thompson KD, Adams A, Richards RH . (2013). Interferon-mediated host response in experimentally induced salmonid alphavirus 1 infection in Atlantic salmon (Salmo salar L.). Vet Immunol Immunopathol 155: 9–20.
Hjortaas MJ, Skjelstad HR, Taksdal T, Olsen AB, Johansen R, Bang-Jensen B et al. (2013). The first detections of subtype 2–related salmonid alphavirus (SAV2) in Atlantic salmon, Salmo salar L., in Norway. J Fish Dis 36: 71–74.
Hodneland K, Bratland A, Christie KE, Endresen C, Nylund A . (2005). New subtype of salmonid alphavirus (SAV), Togaviridae, from Atlantic salmon Salmo salar and rainbow trout Oncorhynchus mykiss in Norway. Dis Aquat Org 66: 113–120.
Houston RD, Taggart JB, Cezard T, Bekaert M, Lowe NR, Downing A et al. (2014). Development and validation of a high density SNP genotyping array for Atlantic salmon (Salmo salar. BMC Genomics 15: 90.
Houston RD, Haley CS, Hamilton A, Guy DR, Tinch AE, Taggart JB et al. (2008). Major quantitative trait loci affect resistance to infectious pancreatic necrosis in Atlantic salmon (Salmo salar. Genetics 178: 1109–1115.
Houston RD, Haley CS, Hamilton A, Guy DR, Mota-Velasco JC, Gheyas AA et al. (2010). The susceptibility of Atlantic salmon fry to freshwater infectious pancreatic necrosis is largely explained by a major QTL. Heredity 105: 318–327.
Houston RD, Davey JW, Bishop SC, Lowe NR, Mota-Velasco JC, Hamilton A et al. (2012). Characterisation of QTL-linked and genome-wide restriction site-associated DNA (RAD) markers in farmed Atlantic salmon. BMC Genomics 13: 244.
Jansen MD, Wasmuth MA, Olsen AB, Gjerset B, Modahl I, Breck O et al. (2010a). Pancreas disease (PD) in sea-reared Atlantic salmon, Salmo salar L., in Norway; a prospective, longitudinal study of disease development and agreement between diagnostic test results. J Fish Dis 33: 723–736.
Jansen MD, Taksdal T, Wasmuth MA, Gjerset B, Brun E, Olsen AB et al. (2010b). Salmonid alphavirus (SAV) and pancreas disease (PD) in Atlantic salmon, Salmo salar L., in freshwater and seawater sites in Norway from 2006 to 2008. J Fish Dis 33: 391–402.
Jansen MD, Jensen BB, Brun E . (2014). Clinical manifestations of pancreas disease outbreaks in Norwegian marine salmon farming – variations due to salmonid alphavirus subtype. J Fish Dis 38: 343–353.
Karlsen M, Tingbo T, Solbakk IT, Evensen O, Furevik A, Aas-Eng A . (2012). Efficacy and safety of an inactivated vaccine against Salmonid alphavirus (family Togaviridae). Vaccine 30: 5688–5694.
Kjøglum S, Henryon M, Aasmundstad T, Korsgaard I . (2008). Selective breeding can increase resistance of Atlantic salmon to furunculosis, infectious salmon anaemia and infectious pancreatic necrosis. Aquac Res 39: 498–505.
Knott SA, Elsen JM, Haley CS . (1996). Methods for multiple-marker mapping of quantitative trait loci in half-sib populations. Theor Appl Genet 93: 71–80.
Knott SA, Haley CS . (1998). Simple multiple-marker sib-pair analysis for mapping quantitative trait loci. Heredity 81: 48–54.
Kristoffersen AB, Viljugrein H, Kongtorp RT, Brun E, Jansen PA . (2009). Risk factors for pancreas disease (PD) outbreaks in farmed Atlantic salmon and rainbow trout in Norway during 2003–2007. Prev Vet Med 90: 127–136.
Li J, Boroevich KA, Koop BF, Davidson WS . (2011). Comparative genomics identifies candidate genes for Infectious Salmon Anemia (ISA) resistance in Atlantic Salmon (Salmo salar. Mar Biotechnol 13: 232–241.
Lien S, Gidskehaug L, Moen T, Hayes BJ, Berg PR, Davidson WS et al. (2011). A dense SNP-based linkage map for Atlantic salmon (Salmo salar reveals extended chromosome homeologies and striking differences in sex-specific recombination patterns. Bmc Genomics 12: 615.
McLoughlin MF, Nelson RN, McCormick JI, Rowley HM, Bryson DB . (2002). Clinical and histopathological features of naturally occurring pancreas disease in farmed Atlantic salmon, Salmo salar L. J Fish Dis 25: 33–43.
Meuwissen T, Goddard M . (1996). The use of marker haplotypes in animal breeding schemes. Genet Sel Evol 28: 161–176.
Moen T, Hayes BJ, Baranski M, Berg PR, Kjoglum S, Koop BF et al. (2008). A linkage map of the Atlantic salmon (Salmo salar based on EST-derived SNP markers. Bmc Genomics 9: 223.
Moen T, Baranski M, Sonesson AK, Kjoglum S . (2009). Confirmation and fine-mapping of a major QTL for resistance to Infectious Pancreatic Necrosis in Atlantic salmon (Salmo salar: population-level associations between markers and trait. BMC Genomics 10: 368.
Nirea K, Sonesson AK, Woolliams J, Meuwissen T . (2012). Strategies for implementing genomic selection in family-based aquaculture breeding schemes: double haploid sib test populations. Genet Sel Evol 44: 30.
Norris A, Foyle L, Ratcliff J . (2008). Heritability of mortality in response to a natural pancreas disease (SPDV) challenge in Atlantic salmon, Salmo salar L., post-smolts on a West of Ireland sea site. J Fish Dis 31: 913–920.
Rastas P, Paulin L, Hanski I, Lehtonen R, Auvinen P . (2013). Lep-MAP: Fast and accurate linkage map construction for large SNP datasets. Bioinformatics 29: 3128–3134.
Rodger H, Mitchell S . (2007). Epidemiological observations of pancreas disease of farmed Atlantic salmon, Salmo salar L., in Ireland. J Fish Dis 30: 157–167.
Sonesson AK, Meuwissen T . (2009). Testing strategies for genomic selection in aquaculture breeding programs. Genet Sel Evol 41: 37.
Stene A, Bang Jensen B, Knutsen Ø, Olsen A, Viljugrein H . (2013). Seasonal increase in sea temperature triggers pancreas disease outbreaks in Norwegian salmon farms. J Fish Dis 37: 739–751.
Storset A, Strand C, Wetten M, Sissel K, Rarnstad A . (2007). Response to selection for resistance against infectious pancreatic necrosis in Atlantic salmon (Salmo salar L.). Aquaculture 272: S62–S68.
Taggart JB . (2007). FAP: An exclusion-based parental assignment program with enhanced predictive functions. Mol Ecol Notes 7: 412–415.
Taksdal T, Olsen AB, Bjerkas I, Hjortaas MJ, Dannevig BH, Graham DA et al. (2007). Pancreas disease in farmed Atlantic salmon, Salmo salar L., and rainbow trout, Oncorhynchus mykiss (Walbaum), in Norway. J Fish Dis 30: 545–558.
Vandeputte M, Mauger S, Dupont-Nivet M . (2006). An evaluation of allowing for mismatches as a way to manage genotyping errors in parentage assignment by exclusion. Mol Ecol Notes 6: 265–267.
Visscher PM, Thompson R, Haley CS . (1996). Confidence intervals in QTL mapping by bootstrapping. Genetics 143: 1013–1020.
Wheatley SB, McLoughlin MF, Menzies FD, Goodall EA . (1995). Site management factors influencing mortality rates in Atlantic salmon (Salmo salar L) during marine production. Aquaculture 136: 195–207.
Acknowledgements
The authors would like to thank Roy Hjelmeland (Marine Harvest, Norway) and Christian Wallace (VESO Vikan, Norway) for clarifications of the challenge protocol, Natalie Lowe (The Roslin Institute, UK) for help with fry tissue sample processing, and John Taggart (Institute of Aquaculture, UK) for assistance with parental assignment. The authors would also like to acknowledge VESO Vikan (POP 1) and PHARMAQ AS (POP 2) for the use of their viral challenge data, and LGC Genomics for genotyping (POP 1). We acknowledge funding from the Biotechnology and Biological Sciences Research Council (BBSRC) (BB/H022007/1), from the Roslin Institute's BBSRC Institute Strategic Funding Grant, and from SalmoBreed AS.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Gonen, S., Baranski, M., Thorland, I. et al. Mapping and validation of a major QTL affecting resistance to pancreas disease (salmonid alphavirus) in Atlantic salmon (Salmo salar). Heredity 115, 405–414 (2015). https://doi.org/10.1038/hdy.2015.37
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2015.37
This article is cited by
-
A high-density genetic map construction and sex-related loci identification in Chinese Giant salamander
BMC Genomics (2021)
-
Genome-Wide Association Analysis Reveals the Genetic Architecture of Parasite (Cryptocaryon irritans) Resistance in Large Yellow Croaker (Larimichthys crocea)
Marine Biotechnology (2021)
-
Identification of genetic loci associated with higher resistance to pancreas disease (PD) in Atlantic salmon (Salmo salar L.)
BMC Genomics (2020)
-
Response of the Salmon Heart Transcriptome to Pancreas Disease: Differences Between High- and Low-Ranking Families for Resistance
Scientific Reports (2020)
-
Genome-wide association mapping and accuracy of predictions for amoebic gill disease in Atlantic salmon (Salmo salar)
Scientific Reports (2020)
