
Abstract
Genomic rearrangements can result in losses, amplifications, translocations and inversions of DNA fragments thereby modifying genome architecture, and potentially having clinical consequences. Many genomic disorders caused by structural variation have initially been uncovered by early cytogenetic methods. The last decade has seen significant progression in molecular cytogenetic techniques, allowing rapid and precise detection of structural rearrangements on a whole-genome scale. The high resolution attainable with these recently developed techniques has also uncovered the role of structural variants in normal genetic variation alongside single-nucleotide polymorphisms (SNPs). We describe how array-based comparative genomic hybridisation, SNP arrays, array painting and next-generation sequencing analytical methods (read depth, read pair and split read) allow the extensive characterisation of chromosome rearrangements in human genomes.
Similar content being viewed by others

Introduction
Diverse types of genomic variants have been described (Scherer et al., 2007) thanks to the development and expansion of molecular biology and cytogenetic techniques, and contribute largely to human disease, normal phenotypic variation and karyotypic evolution. Structural variants (SVs) within individual genomes result from chromosomal rearrangements affecting at least 50 bp (Alkan et al., 2011a) and include deletions and duplications known as copy-number variants (CNVs), inversions and translocations. Rearrangements are triggered by multiple events including external factors such as cellular stress and incorrect DNA repair or recombination (Mani and Chinnaiyan, 2010). Notably, segmental duplications (low-copy repeats), which are particularly frequent in subtelomeric regions (Linardopoulou et al., 2005), facilitate nonallelic homologous recombination and are considered as hotspots for recurrent rearrangements (Mefford and Eichler, 2009; Stankiewicz and Lupski, 2010; Ou et al., 2011).
The conventional cytogenetic methods, ‘chromosome banding’ and ‘karyotyping’ are very informative and are still commonly used. However, these techniques are limited to the detection of numerical chromosomal aberrations (aneuploidy, polyploidy) and microscopic SVs a few megabases in size (Table 1). Molecular cytogenetic approaches enable the detection of submicroscopic SVs and have been crucial for studying complex rearrangements, generated by more than two chromosomal breakage events, refining breakpoints and performing cross-species comparisons (Speicher and Carter, 2005). These newer approaches have mostly relied on the use of ‘fluorescence in situ hybridisation’ (FISH; Bauman et al., 1980) where fluorescence microscopy reveals the presence and localisation of defined labelled DNA probes binding to complementary sequences on targets, traditionally metaphase chromosome spreads. To facilitate detection of events such as translocations, whole chromosome-specific DNA probes or ‘paints’ have been used (‘chromosome painting’; Cremer et al., 1988; Lichter et al., 1988; Pinkel et al., 1988). To increase resolution, shorter probes have been introduced (for example, fosmids and very recently oligonucleotide libraries; Yamada et al., 2011) and/or the target has been refined by replacing condensed chromosomes with extended chromatin fibres (‘Fibre-FISH’; Heng et al., 1992; Wiegant et al., 1992; Parra and Windle, 1993). Furthermore, Fibre-FISH is now facilitated by an automated procedure called ‘molecular combing’ (Michalet et al., 1997). Alternative targeted approaches have simplified CNV detection (Feuk et al., 2006). For example, ‘real-time qPCR’ (Bieche et al., 1998) and ‘MLPA’ (multiplex ligation-dependent probe amplification) are broadly used to detect recurrent events in clinical genetics (Schouten et al., 2002). While these different approaches are restricted to specific regions, some FISH-based techniques have been developed to detect genomic aberrations at the whole-genome level without prior knowledge (Table 1). For example, copy number differences between two genomes can be detected using ‘comparative genomic hybridisation’ (CGH; Kallioniemi et al., 1992); and subtle translocations and complex rearrangements can be characterised using techniques derived from chromosome painting such as ‘M-FISH’ (multiplex-FISH; Speicher et al., 1996) and ‘SKY’ (spectral karyotyping; Schrock et al., 1996) where all chromosomes are differentially coloured in a single experiment (Darai-Ramqvist et al., 2006; Stephens et al., 2011). These methods are experimentally demanding, labour-intensive and the resolution still limited by the use of chromosomes as targets (Table 1).
Precise determination of SV boundaries is crucial for accurate genotype–phenotype correlations, which are dependent on the extent of genes or regulatory regions that are disrupted or vary in copy number (Huang et al., 2010). In addition, nucleotide breakpoint resolution gives insights into the mechanisms underlying SV formation (Korbel et al., 2007; Gu et al., 2008; Kidd et al., 2008; Conrad et al., 2010a; Stankiewicz and Lupski, 2010; Mills et al., 2011). Completion of the human genome sequence in the early 2000s (Lander et al., 2001; Venter et al., 2001) and progress in molecular biology techniques gave rise to new genome-wide screening methods, revolutionising the understanding of the genomes of healthy individuals (Iafrate et al., 2004; Sebat et al., 2004; Redon et al., 2006; Conrad et al., 2010b) as well as patients with disease. In this review, we will discuss how microarray and next-generation sequencing (NGS) technologies can be utilised to reveal and extensively characterise chromosome rearrangements. While the focus of this review is on humans, since the majority of techniques presented here have largely been developed to study human genomes, these new advances are species-independent and hold great promise for future studies in various areas, including karyotype evolution and phylogenomics (Griffin et al., 2008; Skinner et al., 2009; Volker et al., 2010).
Array-based techniques
A brief introduction to arrays
DNA microarrays or ‘chips’ are currently applied to a wide range of applications in molecular biology. Originally developed for gene expression profiling, they are now commonly used to unmask copy number changes (array-based CGH), for single-nucleotide polymorphism (SNP) genotyping, as well as to study DNA methylation, alternative splicing, miRNAs and protein–DNA interactions (array-based ChIP (Chromatin ImmunoPrecipitation)). In short, each array consists of thousands of immobilised nucleic acid sequences (for example, oligonucleotide probes or cloned sequences). Labelled DNA or RNA fragments are applied to the array surface, allowing the hybridisation of complementary sequences between ‘probes’ and ‘targets’. The main advantages of this technology are its sensitivity, specificity and scale as it enables data for thousands of relevant genomic regions of interest to be generated rapidly in a single experiment. Lastly, but important for precious clinical samples, the amount of input sample material required is generally low, usually <1 μg.
CNV discovery using CGH and SNP arrays
While CGH arrays were fabricated specifically for the detection of CNVs in genomes, SNP arrays, initially designed for large-scale genotyping and essential for linkage and association studies, can also be used for this purpose. The genome-wide coverage of features on these arrays allows the discovery of CNVs without any prior knowledge. Some commercial arrays are designed to more easily identify recurrent rearrangements (in particular microdeletion syndromes) or to genotype CNVs present in >1% of the general population (known as copy number polymorphisms, CNP; Alkan et al., 2011a). A list of current commercial human catalogue oligonucleotide arrays is provided in Supplementary Table S1, and arrays are also available for multiple organisms. In addition, array vendors generally provide flexibility in design such that the researcher can easily adapt the content of the array in order to increase the resolution in one or more regions relevant for their study (‘custom designs’).
Array-CGH
The first array-based CGH experiments (Solinas-Toldo et al., 1997; Pinkel et al., 1998) were designed to improve the resolution obtained with conventional CGH (Kallioniemi et al., 1992). Normal metaphase chromosomes were replaced with arrays containing thousands of DNA sequences. Initially, these sequences were large genomic clones of typically 80–200 kb in length, namely BAC or PAC (bacterial/P1-derived artificial chromosome) clones selected throughout the genome at 1-Mb intervals (∼3000 BAC clones per array) (Snijders et al., 2001; Fiegler et al., 2003a; Chung et al., 2004). In 2004, the first whole-genome tiling path array was created (Ishkanian et al., 2004). This array comprised >30 000 overlapping BAC clones covering the entire genome, increasing the array resolution and the potential to detect copy number changes. Array resolution has further improved since technology has allowed an increase in the number of features present on an array and shorter sequences have been used as targets: cDNA (Pollack et al., 1999; Heiskanen et al., 2000), PCR amplicons (Mantripragada et al., 2004; Dhami et al., 2005) and above all, oligonucleotide probes that are now widely used (Brennan et al., 2004; Carvalho et al., 2004). This recent significant increase in array resolution has allowed the detection of genetic imbalances as small as just a few kilobases in size and also enables the boundaries of an imbalance to be better defined.
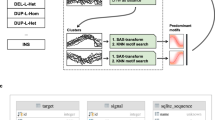
In array-CGH, test and reference DNAs are labelled with different fluorophores (for example, Cy5 and Cy3), and then simultaneously hybridised onto arrays in the presence of Cot-1 DNA to reduce the binding of repetitive sequences (Figure 1). If only low amounts of DNA are available (for example, in prenatal diagnosis or tumour analysis), amplification methods can be applied before labelling (Guillaud-Bataille et al., 2004; Le Caignec et al., 2006; Fiegler et al., 2007) although data quality is in general substantially reduced (Talseth-Palmer et al., 2008; Przybytkowski et al., 2011). After hybridisation, washing and scanning, Cy5 and Cy3 fluorescence intensities are measured for each feature on the array, normalised, and log2 ratios of the test DNA (for example, Cy5) divided by the reference DNA (for example, Cy3) are then plotted against chromosome position. Theoretically, for each position, a value of 0 indicates a normal copy number (log2 (2/2)=0) result, while a log2 ratio of 0.58 (log2 (3/2)=0.58) indicates a one copy gain in test compared with reference, and a log2 ratio of −1 (log2 (1/2)=−1) indicates a one copy loss in test compared with reference. To minimise the influence of CNVs in the reference DNA for the identification of CNVs in the test DNA, a pool of ‘normal’ DNA samples, ideally >100, can be used as a reference. A large variety of algorithms designed to detect CNVs from array-CGH data (‘calling’ algorithms) have been published, for example, ‘DNAcopy’ (Olshen et al., 2004), ‘SW-ARRAY’ (Price et al., 2005), ‘SMAP’ (Andersson et al., 2008), ‘GADA’ (Pique-Regi et al., 2008) and ‘ADM3’ (R package available at http://cran.r-project.org). These algorithms search for intervals where the average log2 ratio exceeds specified thresholds. If probe response is good and background noise is low, a few probes can be sufficient to detect imbalanced regions with confidence (generally a minimum of 3–10 probes are used, depending on platforms; Alkan et al., 2011a). Algorithms can more accurately detect CNVs and will produce less false positive calls if data are normalised to correct for artefacts such as GC-bias, waves (Marioni et al., 2007) or dye-bias (Fitzgerald et al., 2011).

Overview of ‘cytogenetics’ oligonucleotide arrays workflow. White boxes: sample preparation stage, grey boxes: microarray stage. Different methods are available for array-CGH labelling (enzymatic, restriction digestion, Universal Linkage System) and can require a fragmentation step (dashed line box). Hybridisation mixtures contain blocking agents and DNA enriched for repetitive sequences (for example, Cot-1 DNA) to block nonspecific hybridisation and reduce background signal. Hybridisation times vary according to platform and array format. For further details on protocols see the commercial vendors' website. Available catalogue arrays are listed in Supplementary Table S1. Cy5, cyanine-5; Cy3, cyanine-3; gDNA, genomic DNA; OGT, Oxford Gene Technology; WGA, whole-genome amplification.
Commercial arrays (Supplementary Table S1) provided by companies such as Agilent Technologies (Santa Clara, CA, USA) BlueGnome (Cambridge, UK) Oxford Gene Technology (Oxford, UK) and Roche NimbleGen (Madison, WI, USA) in the UK, offer robustness, sensitivity and flexibility compared with early BAC arrays. As previously stated, the researcher can order a custom design including dense coverage focusing on single or multiple chromosomal regions where higher resolution is required. Conrad et al. (2010b) describe the use of a set of 20 ultra-high resolution oligonucleotide arrays comprising 42 million probes in total, with a median probe spacing of just 56 bp across the entire genome. Such high resolution enabled the identification of 11 700 CNVs >443 bp in the genomes of 40 normal individuals. The fabrication processes of the arrays vary between manufacturers. For example, Agilent Technologies utilises in situ inkjet technology (‘SurePrint Technology’, Agilent Technologies) to synthesise 60-mer oligonucleotide array features (Barrett et al., 2004). This technology produces highly reproducible features and excellent signal-to-noise ratios, assuring maximum sensitivity and specificity. Custom arrays can be designed and ordered using the online eArray application (https://earray.chem.agilent.com/earray/), which contains at present over 28 million in silico-validated human oligonucleotide sequences. These 60-mers span exonic, intronic, intergenic, pseudoautosomal, segmented duplication DNA regions and copy number variable regions. In addition to sequences contained in the database, any custom oligonucleotide sequence with a size ranging from 25 to 60 bp can be printed. For every oligonucleotide on the array, scores can be provided by array manufacturers, which can predict their performance on a genomic array and help to interpret derivative log2 ratio values in breakpoint regions (Sharp et al., 2007). Scores are based on various parameters such as melting temperature (Tm), SNP content, sequence complexity and uniqueness of the oligonucleotide sequence. For cost-effectiveness, the user can choose between different layouts, from 8 × 60 K up to 1 × 1 M for SurePrint G3 arrays (Supplementary Table S1). Furthermore, designs can be shared with collaborators through the online application. Roche NimbleGen high-density array manufacturing is based on photo-mediated synthesis process using the Maskless Array Synthesizer technology (Nuwaysir et al., 2002). In comparison to other in situ synthesis technologies such as inkjet deposition, this method enables the production of more features on the glass slide, and oligonucleotide lengths are usually ranging between 50 and 75 bp. They have recently introduced very high-resolution arrays composed of 4.2 million array features (284 bp median feature spacing), and different array formats are also available (Supplementary Table S1). The array design is made on-demand by Roche NimbleGen from a list of regions of interest supplied by the customer. Similar to Agilent Technologies, Roche NimbleGen offers whole-genome catalogue arrays and custom solutions designed to study a range of various organisms.
SNP arrays
As with array-CGH technology, SNP arrays have undergone huge developments over the last few years (Kennedy et al., 2003; Gunderson et al., 2005; LaFramboise, 2009), with the ability to genotype a few thousands SNPs at first, rising to millions of SNPs today in the latest arrays. In addition to the advances in resolution, the design of the arrays is continually incorporating more informative SNPs, as a result of large-scale studies such as the HapMap Project (The International HapMap Consortium, 2003) and the 1000 genomes project (Durbin et al., 2010). Although SNPs account for a substantial part of genetic variation, chromosomal rearrangements have a tremendous role in disease, evolution and tumourigenesis, and SNP arrays have progressively started to be used to simultaneously genotype SNPs and detect rare and common genomic rearrangements (Bignell et al., 2004; Huang et al., 2004; Peiffer et al., 2006). Besides amplifications and deletions detected by both CGH and SNP arrays, SNP arrays can reveal mosaicism, extended regions of loss of heterozygosity, uniparental disomy (Conlin et al., 2010), provide more accurate calculation of copy numbers (Greenman et al., 2010) and determine parental origin of de novo CNVs (Conlin et al., 2010) in trios. Unlike array-CGH, which relies on co-hybridisation of test and reference DNA, only the test sample is hybridised onto each SNP array (Figure 1). The copy-number analysis of SNP array data generally uses two parameters, comparing observed test sample values to expected reference values, the Log2 R intensity ratio, and the allelic intensity ratio or ‘B-allele frequency’ (Peiffer et al., 2006; Alkan et al., 2011a). Many algorithms have been developed and are often specific to array types (Winchester et al., 2009; Dellinger et al., 2010; Pinto et al., 2011). To improve the efficiency of CNV discovery with SNP arrays, manufacturers have included nongenotyping, nonpolymorphic markers in their designs, which are specifically designed to detect CNVs with greater performance, as well as increasing marker density in CNV regions (Supplementary Table S1). For example, half of the 1.8 million markers of the human Affymetrix 6.0 array are dedicated to the identification of copy-number variation (McCarroll et al., 2008).
Should SNP arrays replace CGH arrays? Despite the variety of information obtained in a single experiment, greater potential for automation and scalability, SNP arrays generally do not perform as well as dedicated CGH arrays for copy-number variation discovery, in terms of sensitivity and resolution (Cooper et al., 2008; Curtis et al., 2009; Alkan et al., 2011a; Pinto et al., 2011). To conclude, the choice of platform should be dependent on the project. If looking for very small deletions (<50 kb) or gains, array-CGH would probably be the best option. However, for cancer genetics or human diseases linked to uniparental disomy, for example, Prader–Willi and Angelman syndromes (Yamazawa et al., 2010), SNP arrays could be more appropriate. Recently, several companies have been developing hybrid arrays designed both for copy-number analysis and for detection of mosaicism, loss of heterozygosity, uniparental disomy or regions identical by descent, using allelic difference features (‘CGH+SNP’ array and ‘cytogenetics’ array) (Figure 1; Supplementary Table S1). However, the performance of these platforms is not widely reported to date and they have not yet been included in platform comparison studies.
Fine-mapping of translocation breakpoints using array painting
Although array-CGH can be used to reveal deletions and amplifications, including imbalances associated with apparently balanced translocation, they are unable to detect balanced rearrangement events such as inversions and balanced reciprocal translocations. Balanced reciprocal translocations are carried constitutionally by 1 in 500 individuals and also occur frequently in cancer cells (Howarth et al., 2008). Disruption of regulatory regions such as enhancers or genes, and creation of fusion transcripts by a chromosome translocation can have phenotypic consequences. In this section, we will describe the ‘array painting’ technique, which combines flow-sorting of derivative chromosomes and array-CGH to map translocation breakpoints and identifies more accurately gene disruption.
Array painting is a technique derived from reverse chromosome painting (Carter et al., 1992) and array-CGH technologies, developed to rapidly characterise reciprocal chromosome translocation breakpoints (Fiegler et al., 2003b) (Figure 2). In reverse chromosome painting, probes are generated by DOP-PCR (degenerate oligonucleotide primed PCR; Telenius et al., 1992) from isolated aberrant chromosomes, and hybridised onto normal metaphase spreads using FISH. This enables the identification of chromosomal regions present in the aberrant chromosome, and to locate the approximate positions of the breakpoints. As with conventional CGH (Kallioniemi et al., 1992), using metaphase chromosomes as a target limits the resolution of reverse painting and breakpoints can only be localised at a resolution of 5–10 Mb. In order to increase accuracy, metaphase chromosomes have been replaced by arrays (Fiegler et al., 2003b).

Overview of array painting workflow. White boxes: sample preparation stage, grey boxes: microarray stage. BAC, bacterial artificial chromosome; WGA, whole-genome amplification. For further details see Gribble et al. (2009).
First, the two aberrant or ‘derivative’ chromosomes involved in the reciprocal translocation are isolated. This can be achieved by flow-sorting (Gribble et al., 2009) or by microdissection (Backx et al., 2007). Subsequently, each derivative chromosome, represented by one (Gribble et al., 2004) or generally multiple copies, is amplified using DOP-PCR or commercially available whole-genome amplification kits to provide sufficient DNA. The amplified products are then differentially labelled with fluorescent dyes (Cy5 and Cy3), and co-hybridised onto an array, which is then scanned after excess labelled probe is washed off (Figure 2). As for array-CGH, log2 ratios for Cy5/Cy3 intensities are plotted against chromosome position for each feature. Because the chromosomal regions flanking each side of the breakpoint are differentially labelled as they are present on different derivative chromosomes, the position where the log2 ratios changes from high to low ratios (or vice versa) defines the breakpoint, and breakpoint spanning clones usually show intermediate ratios (Fiegler et al., 2003b; Backx et al., 2007) (Figure 2). Fine-mapping of breakpoints is only dependent on the resolution of the array. In the initial reports of the array painting method, 1-Mb whole-genome or custom tiling BAC arrays were used (Fiegler et al., 2003b). Array painting benefited from the evolution of array-CGH technology and BAC arrays have been replaced by whole-genome or region-specific high-resolution oligonucleotide arrays, allowing higher resolution and better accuracy of breakpoint determination (Gribble et al., 2007). Precise breakpoint mapping of balanced translocations can give insights into associated phenotypes in patients. For example, array painting performed with a 244 K CGH array for a t(10;13)(q22;p13) balanced translocation suggested that C10orf11, which was disrupted by the translocation, could contribute to the mental retardation phenotype in 10q22 deletion patients (Tzschach et al., 2010). Breakpoints identified by array technologies can be independently validated by FISH assays to visually demonstrate the rearrangements in individual cells.
This robust procedure can be used to determine the composition of any isolated chromosome and has applications other than mapping balanced translocation breakpoints. Thus, complex chromosome rearrangements, involving more than two chromosomes, can be deciphered (Fauth et al., 2006), and in some instances other inter-chromosomal aberrations may be identified. Furthermore, array painting can replace conventional chromosome painting to determine cross-species homology, which can give insights into karyotype evolution. For example, white-cheeked gibbon chromosome 14 was hybridised onto a human 1-Mb array, which identified syntenic blocks on human chromosomes 2 and 17 (Gribble et al., 2004).
An alternative technique to array painting for fine-mapping of translocation and complex rearrangements breakpoints, based on ‘Chromatin Conformation Capture on Chip’ or 4C (Simonis et al., 2009) has been described. Briefly, many fragments across the breakpoints are captured by cross-linking physically close parts of the genome, followed by restriction enzyme digestion, locus-specific inverse PCR and templates hybridised to 4C-tailored microarrays. Clustering of positive signals displaying increased intensities predicts breakpoints at the resolution of the array. It claims to be particularly valuable if isolation of derivative chromosomes is not achievable, and to characterise inversions.
NGS-based techniques
A brief introduction to NGS
Using conventional Sanger sequencing, it has taken more than a decade of international effort to sequence the human genome (Lander et al., 2001; Venter et al., 2001). Since the development of NGS (or ‘second-generation’ sequencing) technologies in 2005, sequencing of a whole human genome can now be achieved in a few days and at much lower cost. Also known as ‘massively parallel sequencing’, these technologies allow the sequencing of millions of DNA molecules simultaneously after library preparation of fragments, to produce sequence reads. Sequence reads are generally aligned to the reference genome and base variants, small insertions/deletions (indels) and SVs (>50 bp) can be detected. The most commonly used platforms at present have been developed by Illumina (Genome Analyzer/HiSeq, San Diego, CA, USA), Roche (454 Life Sciences, Branford, CT, USA) and Applied Biosystems/Life Technologies (SOLiD, Foster City, CA, USA) and these as well as others are reviewed by Metzker (2010). In addition to high-throughput resequencing for understanding human genome variation and diseases, this technology has opened the door to a wide range of applications such as large-scale gene expression studies using RNAseq, and whole-genome sequencing of many organisms, which has a huge impact on evolutionary knowledge. NGS technologies are still under development and third-generation platforms could produce reads reaching up to a few kilobases whereas read lengths presently range from ∼30 to ∼400 bp depending on the platform (Metzker, 2010). Until whole-genome sequencing becomes more economical, specific genomic regions can be isolated for sequencing, for example, chromosomes or derivative chromosomes can be isolated by flow-sorting, or regions of interest can be selected from the genome by sequence capture (also termed ‘pull-down’ or ‘enrichment’; Coffey et al., 2011; Hedges et al., 2011). Another way to make NGS more cost-effective when working with small genomes or specific genomic regions is to add a unique oligonucleotide ‘tag’ or ‘index’ to samples before multiplexing and sequencing (Parameswaran et al., 2007).
Deciphering chromosomal rearrangements with NGS technology
Information provided by read mapping and sequence coverage enables the detection of SVs and NGS is becoming an attractive alternative to array-based assays in the field of molecular cytogenetics. Among the many advantages of high-throughput NGS, SVs of all types and sizes can theoretically be detected, breakpoints can be mapped with high resolution, down to the basepair level in some instances, and complex rearrangements can be characterised with the possibility to study multiple breakpoints in a single experiment. Four different approaches have been described to characterise SVs: (i) read-depth analysis, which can only detect gains and losses; (ii) read-pair analysis (paired-end mapping); (iii) split-read analysis; and (iv) assembly methods, all of which can detect in theory all types of rearrangements including copy-neutral rearrangements (inversions and translocations) (Figure 3). A variety of tools based on one or more of these methods have been developed to analyse chromosomal rearrangements according to the genomic regions affected, the size-range and breakpoint precision (Medvedev et al., 2009; Alkan et al., 2011a; Mills et al., 2011). We will discuss how each method can be used to characterise genomic rearrangements, with the exception of local assembly approaches that are still limited by read length and cost (Alkan et al., 2011b).

Four methods to identify SVs from NGS data. These methods are often used in combination to detect chromosomal rearrangements and characterise breakpoints (red arrows) with precision. De novo assembly methods are still challenging but have the potential to accurately and rapidly characterise all classes of rearrangements. MEI, mobile-element insertion; RP, read pair. For further details and full figure legend, see Alkan et al., 2011a. Reprinted by permission from Macmillan Publishers Ltd: Nature Reviews Genetics (Alkan et al., 2011a), copyright 2011.
Read-depth method
Read-depth NGS data (Campbell et al., 2008; Chiang et al., 2009; Yoon et al., 2009) are essentially providing similar information to that obtained from array-CGH, by indicating copy-number gains (>2 copies for a diploid genome) or losses (<2 copies). Sequence read depth, that is, the number of reads mapping at each chromosomal position, is in theory randomly dispersed and significant divergence from the normal Poisson distribution indicates copy-number variation (Figure 3). Duplications and amplifications are indicated by the presence of regions showing excessive read depth, whereas low read depth indicates heterozygous deletion and absence of coverage is suggestive of homozygous deletion. Statistical power is limited for smaller CNVs but increasing sequence coverage can in some instances improve sensitivity (Chiang et al., 2009). Factors such as GC content, homopolymeric stretches of DNA or preferential PCR amplification at the library preparation stage can introduce biases. Repetitive DNA regions are also problematic as reads are aligned with low confidence (low ‘mapping quality’; Li et al., 2008), providing poor information on copy-number status, but longer reads will increase mapping specificity in the future. Applying read-depth analysis to cancer cell lines has shown that the dynamic range for absolute copy-number evaluation is greater than that detected by SNP arrays (Campbell et al., 2008), which tend to saturate for high intensity values. For example, Chiang et al. (2009) found a 55.6-fold increase by NGS compared with only a 16-fold increase by SNP array for the ERBB2 locus in a breast carcinoma cell line. This increased dynamic range of NGS may lead to new insights into segmental duplications (Alkan et al., 2009) and multicopy gene families (Sudmant et al., 2010).
Read-pair method
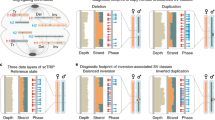
Currently, the most powerful method to study chromosome rearrangements is the paired-end read mapping technique (Tuzun et al., 2005; Korbel et al., 2007) (Figure 3). Sequence read pairs are short sequences from both ends of each of the millions of DNA fragments (‘inserts’) generated by library preparation. Clustering of at least two discordant pairs of reads, either by size or by orientation, is suggestive of a chromosome rearrangement. When aligned to the reference genome, read pairs (><) are expected to map at a certain distance (>----< → >----<) corresponding to the average library insert size (typically 200–500 bp and up to 5 kb for large-insert libraries); a spanning distance significantly different from the average insert size indicates putative SVs. Deletions are identified by read pairs spanning a longer genomic region when mapped to the reference not carrying the deletion (>--(del)--< → >--------<). By contrast, insertions or tandem duplications in the sequenced sample will cause the reads to map closer as they are absent from the reference genome (>-(ins/dup)-< → >--<). In addition to the expected span distance of a sequence read pair, aberrant mapping orientation can identify inversions (>---->) and tandem duplications (<---->) (Korbel et al., 2007; Kidd et al., 2008) (Figure 3). Novel insertions, as compared with published reference genomes, are identifiable when only one read of the pair is mapping (<----).
Data from short-insert libraries often need to be supplemented by data from large-insert libraries generated by large circular fragments of DNA typically of 2–5 kb, providing higher physical coverage at breakpoints thereby facilitating SV detection (Shendure et al., 2005; Bentley et al., 2008) (Figure 4). Short-insert libraries (200–500 bp) have a limited capacity to detect SVs mediated by segmental duplications (or low-copy repeats) that harbour a substantial part of SVs (Sharp et al., 2005; Cooper et al., 2007; Kidd et al., 2008; Conrad et al., 2010a), because reads map to multiple similar genomic locations (Li et al., 2008). Another example is the limit to detect insertions larger than the library insert size. Conversely, small events (<400 bp) can be missed with large-insert libraries because the expected size variance between the mate pairs will not be significantly altered. The lower resolution associated with large-insert libraries can also mistake complex events, where several breakpoints are in close proximity such as small inversions flanked by deletions, for simple deletions (Bentley et al., 2008).

Mapping translocation breakpoints by NGS. Bars depict sequencing reads mapping to distinct chromosomes (chromosome 1 and chromosome 2) each side of the translocation breakpoint. Sequence coverage (number of times the breakpoint is covered by sequencing reads) vs physical coverage (number of times the breakpoint is covered by library fragments) are indicated. (a) Single-end sequencing. (b) Paired-end sequencing from a short-insert library (<500 bp). (c) Paired-end sequencing from a large-insert library (>1 kb), increasing physical coverage at the breakpoint site and likelihood of characterising the translocation. Reads spanning the translocation breakpoint are called ‘split reads’ and can identify breakpoints at basepair resolution. Higher depth of sequence coverage (using short-insert libraries) and longer read lengths theoretically generates more informative split reads. Reprinted by permission from Macmillan Publishers Ltd: Nature Reviews Genetics (Meyerson et al., 2010), copyright 2010.
Split-read method
The third approach commonly applied to NGS data is the split-read method (Figure 3). Although this method was originally developed for Sanger sequencing (Mills et al., 2006) and will be much more efficient with longer read length, it is already capable of precisely mapping breakpoints for small deletions (1 bp–10 kb) in unique regions of the genome using the algorithm Pindel (Ye et al., 2009) and read lengths as low as 36 bp. The first stage is to map all reads to the reference genome, and select read pairs responding to the following criteria: one read maps perfectly (no mismatches) and uniquely (no other genomic location), and the other read of the pair cannot be mapped (that is, it is across the rearrangement breakpoint). For each of these pairs, using the location and orientation of the mapped read, Pindel searches for the paired unmapped read (‘split’ read) by performing multiple local alignments. In the case of deletions, candidate unmapped reads are split into two fragments that map separately, and analysis of the alignment deciphers the breakpoint at the basepair level. The AGE algorithm described recently has been designed to identify exact breakpoints for tandem duplications, inversions and complex events (Abyzov and Gerstein, 2011). Thus, this method has a significant advantage on others applied to array or NGS data, which can identify breakpoints with high resolution (Gribble et al., 2007; Mills et al., 2011) but require an additional PCR or high-throughput capture step (Conrad et al., 2010a; Mills et al., 2011) followed by conventional or NGS to reach basepair resolution.
Fine-mapping of translocation breakpoints using NGS
In one of the first studies applying NGS to fine map a reciprocal translocation breakpoint, derivative chromosomes were isolated by flow-sorting, sequenced and single-end reads aligned to the two corresponding chromosomes (Figure 4a). Read-depth analysis identified breakpoints within 1 kb, which were subsequently confirmed at the basepair level by PCR amplification and sequencing (Chen et al., 2008). With whole-genome sequencing becoming more affordable and paired-end technology now available, flow-sorting of derivative chromosomes becomes less critical, as essentially, pairs of reads mapping to different chromosomes will identify translocations (Figures 4b and c; Slade et al., 2010). Large-insert paired-end libraries (Figure 4c) of ∼3 kb are generally preferred to short-insert libraries to increase physical coverage, and maximise chances of observing read pairs consistently spanning the breakpoint (Chen et al., 2010; Slade et al., 2010). If high sequence coverage is reached and reads span the breakpoint (‘split’ reads) (Figure 4), it should be straightforward to directly identify the exact breakpoint without the need for an extra PCR/sequencing step. For example, a method called SLOPE can rapidly identify sequence breakpoints for translocations using read-depth and split-read data (Abel et al., 2010).
Insights from cancer genomes
NGS has also revolutionised the understanding of cancer genomes by identifying not only the full spectrum of somatic point mutations (Mardis et al., 2009; Pleasance et al., 2010) but also giving more insights into complex whole-genome acquired rearrangements (Campbell et al., 2008; Stephens et al., 2009) (Figure 5). These studies showed that intra- and inter-chromosomal somatic rearrangements can be detected and are more frequent than envisaged, partly because they involve small aberrations beyond the resolution of previous molecular cytogenetics methods, emphasising the utility of NGS to study rearrangements (Meyerson et al., 2010). Discovery of fusion genes resulting from these rearrangements and having potential functional consequences is greatly facilitated. Furthermore, transcriptome sequencing using next-generation technologies can identify or validate putative fusion transcripts in a high-throughput manner (Maher et al., 2009).

Genomic landscape of rearrangements in a pancreatic cancer patient. NGS identified various types of inter- and intra-chromosomal rearrangements scattered across the whole genome as shown by this circos plot. Inner ring represents copy-number status and outer ring shows chromosome ideograms. Reprinted by permission from Macmillan Publishers Ltd: Nature (Campbell et al., 2010), copyright 2010.
Impact on present and future studies
Recently developed molecular cytogenetic methods have provided new tools to accurately characterise chromosomal rearrangements and have uncovered the great complexity of human genome architecture (Pang et al., 2010). We have shown that each strategy has limitations, emphasising that approaches often need to be combined to capture the entire range of genetic variation (Alkan et al., 2011a; Mills et al., 2011).
Despite the enormous potential of high-throughput sequencing, array technology has progressed in the past few years and is still appropriate for a broad range of research projects. In addition to robustness, flexibility, and low input material required, array technologies do not demand as many resources as NGS technologies in terms of equipment and computational power. Arrays also give the possibility to study a large number of samples in a cost-effective manner. For example, CNVs identified in discovery phases can be subsequently genotyped by arrays in large population samples and used in disease association studies (Craddock et al., 2010), however data can be less accurate than sequencing at high-copy number states (Chiang et al., 2009). Array-based assays have replaced karyotyping for the diagnosis of developmental disabilities or congenital anomalies (Miller et al., 2010), and will remain the gold standard method until sequencing costs drop dramatically and downstream analyses are facilitated.
Array-based methods have revealed an unexpected level of rearrangement complexity such as imbalances in apparent balanced translocations (Gribble et al., 2005; Howarth et al., 2011), but they are mostly restricted to the detection of CNVs, and FISH is still required to distinguish tandem from dispersed duplications and decode complex rearrangements. Moreover, resolution achieved using arrays can be limited by the density of features printed on the glass slide and there has clearly been a bias towards detecting larger events thus far, even if sets of custom arrays can be employed to increase resolution (Conrad et al., 2010b; Park et al., 2010). The emergence of techniques based on high-throughput sequencing is opening new perspectives for chromosome rearrangement analyses. Whole-genome sequencing is comprehensive and reveals point mutations, indels, as well as all types of chromosome rearrangements including balanced events, and can be used to reconstruct genome architecture. Success of sequencing approaches is often dependent on obtaining sufficient coverage because of the relatively high level of sequencing error in NGS. Current analytical methods mostly rely on sequence alignment against a unique reference genome, and unspecific mapping of short reads to repetitive regions is problematic with many events mediated by repetitive elements potentially being missed (Conrad et al., 2010a). However, third-generation sequencing technologies (Metzker, 2010) will provide longer reads more cheaply, enabling accurate de novo assembly and will help to overcome these issues.
With the increase in resolution and the larger number of SVs detected in each genome with current methods, the challenge is now to infer their phenotypic impact on normal variation and health (Huang et al., 2010). More resources will be needed to guide the interpretation, especially with the growing interest for personalised medicine. Up until now, NGS technologies have largely been applied to study the human genome, but complete sequencing of more than a thousand organisms (997 prokaryotes and 39 eukaryotes, May 2011; http://www.ncbi.nlm.nih.gov/genomes/static/gpstat.html) has now been completed and hundreds more are in progress. Methods described in this review can be utilised to detect and comprehend SV between species or strains and give new insights into recent evolution.
Data Archiving
There was no data to deposit.
References
Abel HJ, Duncavage EJ, Becker N, Armstrong JR, Magrini VJ, Pfeifer JD (2010). SLOPE: a quick and accurate method for locating non-SNP structural variation from targeted next-generation sequence data. Bioinformatics 26: 2684–2688.
Abyzov A, Gerstein M (2011). AGE: defining breakpoints of genomic structural variants at single-nucleotide resolution, through optimal alignments with gap excision. Bioinformatics 27: 595–603.
Alkan C, Coe BP, Eichler EE (2011a). Genome structural variation discovery and genotyping. Nat Rev Genet 12: 363–376.
Alkan C, Kidd JM, Marques-Bonet T, Aksay G, Antonacci F, Hormozdiari F et al. (2009). Personalized copy number and segmental duplication maps using next-generation sequencing. Nat Genet 41: 1061–1067.
Alkan C, Sajjadian S, Eichler EE (2011b). Limitations of next-generation genome sequence assembly. Nat Methods 8: 61–65.
Andersson R, Bruder CE, Piotrowski A, Menzel U, Nord H, Sandgren J et al. (2008). A segmental maximum a posteriori approach to genome-wide copy number profiling. Bioinformatics 24: 751–758.
Backx L, Van Esch H, Melotte C, Kosyakova N, Starke H, Frijns JP et al. (2007). Array painting using microdissected chromosomes to map chromosomal breakpoints. Cytogenet Genome Res 116: 158–166.
Barrett MT, Scheffer A, Ben-Dor A, Sampas N, Lipson D, Kincaid R et al. (2004). Comparative genomic hybridization using oligonucleotide microarrays and total genomic DNA. Proc Natl Acad Sci USA 101: 17765–17770.
Bauman JG, Wiegant J, Borst P, van Duijn P (1980). A new method for fluorescence microscopical localization of specific DNA sequences by in situ hybridization of fluorochromelabelled RNA. Exp Cell Res 128: 485–490.
Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG et al. (2008). Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456: 53–59.
Bieche I, Olivi M, Champeme MH, Vidaud D, Lidereau R, Vidaud M (1998). Novel approach to quantitative polymerase chain reaction using real-time detection: application to the detection of gene amplification in breast cancer. Int J Cancer 78: 661–666.
Bignell GR, Huang J, Greshock J, Watt S, Butler A, West S et al. (2004). High-resolution analysis of DNA copy number using oligonucleotide microarrays. Genome Res 14: 287–295.
Brennan C, Zhang Y, Leo C, Feng B, Cauwels C, Aguirre AJ et al. (2004). High-resolution global profiling of genomic alterations with long oligonucleotide microarray. Cancer Res 64: 4744–4748.
Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA et al. (2010). The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 467: 1109–1113.
Campbell PJ, Stephens PJ, Pleasance ED, O'Meara S, Li H, Santarius T et al. (2008). Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat Genet 40: 722–729.
Carter NP, Ferguson-Smith MA, Perryman MT, Telenius H, Pelmear AH, Leversha MA et al. (1992). Reverse chromosome painting: a method for the rapid analysis of aberrant chromosomes in clinical cytogenetics. J Med Genet 29: 299–307.
Carvalho B, Ouwerkerk E, Meijer GA, Ylstra B (2004). High resolution microarray comparative genomic hybridisation analysis using spotted oligonucleotides. J Clin Pathol 57: 644–646.
Chen W, Kalscheuer V, Tzschach A, Menzel C, Ullmann R, Schulz MH et al. (2008). Mapping translocation breakpoints by next-generation sequencing. Genome Res 18: 1143–1149.
Chen W, Ullmann R, Langnick C, Menzel C, Wotschofsky Z, Hu H et al. (2010). Breakpoint analysis of balanced chromosome rearrangements by next-generation paired-end sequencing. Eur J Hum Genet 18: 539–543.
Chiang DY, Getz G, Jaffe DB, O′Kelly MJ, Zhao X, Carter SL et al. (2009). High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat Methods 6: 99–103.
Chung YJ, Jonkers J, Kitson H, Fiegler H, Humphray S, Scott C et al. (2004). A whole-genome mouse BAC microarray with 1-Mb resolution for analysis of DNA copy number changes by array comparative genomic hybridization. Genome Res 14: 188–196.
Coffey AJ, Kokocinski F, Calafato MS, Scott CE, Palta P, Drury E et al. (2011). The GENCODE exome: sequencing the complete human exome. Eur J Hum Genet 19: 827–831.
Conlin LK, Thiel BD, Bonnemann CG, Medne L, Ernst LM, Zackai EH et al. (2010). Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum Mol Genet 19: 1263–1275.
Conrad DF, Bird C, Blackburne B, Lindsay S, Mamanova L, Lee C et al. (2010a). Mutation spectrum revealed by breakpoint sequencing of human germline CNVs. Nat Genet 42: 385–391.
Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y et al. (2010b). Origins and functional impact of copy number variation in the human genome. Nature 464: 704–712.
Cooper GM, Nickerson DA, Eichler EE (2007). Mutational and selective effects on copy-number variants in the human genome. Nat Genet 39: S22–S29.
Cooper GM, Zerr T, Kidd JM, Eichler EE, Nickerson DA (2008). Systematic assessment of copy number variant detection via genome-wide SNP genotyping. Nat Genet 40: 1199–1203.
Craddock N, Hurles ME, Cardin N, Pearson RD, Plagnol V, Robson S et al. (2010). Genome-wide association study of CNVs in 16 000 cases of eight common diseases and 3000 shared controls. Nature 464: 713–720.
Cremer T, Lichter P, Borden J, Ward DC, Manuelidis L (1988). Detection of chromosome aberrations in metaphase and interphase tumor cells by in situ hybridization using chromosome-specific library probes. Hum Genet 80: 235–246.
Curtis C, Lynch AG, Dunning MJ, Spiteri I, Marioni JC, Hadfield J et al. (2009). The pitfalls of platform comparison: DNA copy number array technologies assessed. BMC Genomics 10: 588.
Darai-Ramqvist E, Diaz de Stahl T, Sandlund A, Mantripragada K, Klein G, Dumanski J et al. (2006). Array-CGH and multipoint FISH to decode complex chromosomal rearrangements. BMC Genomics 7: 330.
Dellinger AE, Saw SM, Goh LK, Seielstad M, Young TL, Li YJ (2010). Comparative analyses of seven algorithms for copy number variant identification from single nucleotide polymorphism arrays. Nucleic Acids Res 38: e105.
Dhami P, Coffey AJ, Abbs S, Vermeesch JR, Dumanski JP, Woodward KJ et al. (2005). Exon array CGH: detection of copy-number changes at the resolution of individual exons in the human genome. Am J Hum Genet 76: 750–762.
Durbin RM, Abecasis GR, Altshuler DL, Auton A, Brooks LD, Durbin RM et al. (2010). A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073.
Fauth C, Gribble SM, Porter KM, Codina-Pascual M, Ng BL, Kraus J et al. (2006). Micro-array analyses decipher exceptional complex familial chromosomal rearrangement. Hum Genet 119: 145–153.
Feuk L, Carson AR, Scherer SW (2006). Structural variation in the human genome. Nat Rev Genet 7: 85–97.
Fiegler H, Carr P, Douglas EJ, Burford DC, Hunt S, Scott CE et al. (2003a). DNA microarrays for comparative genomic hybridization based on DOP-PCR amplification of BAC and PAC clones. Genes Chromosomes Cancer 36: 361–374.
Fiegler H, Geigl JB, Langer S, Rigler D, Porter K, Unger K et al. (2007). High resolution array-CGH analysis of single cells. Nucleic Acids Res 35: e15.
Fiegler H, Gribble SM, Burford DC, Carr P, Prigmore E, Porter KM et al. (2003b). Array painting: a method for the rapid analysis of aberrant chromosomes using DNA microarrays. J Med Genet 40: 664–670.
Fitzgerald TW, Larcombe LD, Le Scouarnec S, Clayton S, Rajan D, Carter NP et al. (2011). aCGH—Spline—An R package for aCGH dye bias normalisation. Bioinformatics 27: 1195–1200.
Greenman CD, Bignell G, Butler A, Edkins S, Hinton J, Beare D et al. (2010). PICNIC: an algorithm to predict absolute allelic copy number variation with microarray cancer data. Biostatistics 11: 164–175.
Gribble SM, Fiegler H, Burford DC, Prigmore E, Yang F, Carr P et al. (2004). Applications of combined DNA microarray and chromosome sorting technologies. Chromosome Res 12: 35–43.
Gribble SM, Kalaitzopoulos D, Burford DC, Prigmore E, Selzer RR, Ng BL et al. (2007). Ultra-high resolution array painting facilitates breakpoint sequencing. J Med Genet 44: 51–58.
Gribble SM, Ng BL, Prigmore E, Fitzgerald T, Carter NP (2009). Array painting: a protocol for the rapid analysis of aberrant chromosomes using DNA microarrays. Nat Protoc 4: 1722–1736.
Gribble SM, Prigmore E, Burford DC, Porter KM, Ng BL, Douglas EJ et al. (2005). The complex nature of constitutional de novo apparently balanced translocations in patients presenting with abnormal phenotypes. J Med Genet 42: 8–16.
Griffin DK, Robertson LB, Tempest HG, Vignal A, Fillon V, Crooijmans RP et al. (2008). Whole genome comparative studies between chicken and turkey and their implications for avian genome evolution. BMC Genomics 9: 168.
Gu W, Zhang F, Lupski JR (2008). Mechanisms for human genomic rearrangements. Pathogenetics 1: 4.
Guillaud-Bataille M, Valent A, Soularue P, Perot C, Inda MM, Receveur A et al. (2004). Detecting single DNA copy number variations in complex genomes using one nanogram of starting DNA and BAC-array CGH. Nucleic Acids Res 32: e112.
Gunderson KL, Steemers FJ, Lee G, Mendoza LG, Chee MS (2005). A genome-wide scalable SNP genotyping assay using microarray technology. Nat Genet 37: 549–554.
Hedges DJ, Guettouche T, Yang S, Bademci G, Diaz A, Andersen A et al. (2011). Comparison of three targeted enrichment strategies on the SOLiD sequencing platform. PLoS One 6: e18595.
Heiskanen MA, Bittner ML, Chen Y, Khan J, Adler KE, Trent JM et al. (2000). Detection of gene amplification by genomic hybridization to cDNA microarrays. Cancer Res 60: 799–802.
Heng HH, Squire J, Tsui LC (1992). High-resolution mapping of mammalian genes by in situ hybridization to free chromatin. Proc Natl Acad Sci USA 89: 9509–9513.
Howarth KD, Blood KA, Ng BL, Beavis JC, Chua Y, Cooke SL et al. (2008). Array painting reveals a high frequency of balanced translocations in breast cancer cell lines that break in cancer-relevant genes. Oncogene 27: 3345–3359.
Howarth KD, Pole JC, Beavis JC, Batty EM, Newman S, Bignell GR et al. (2011). Large duplications at reciprocal translocation breakpoints that might be the counterpart of large deletions and could arise from stalled replication bubbles. Genome Res 21: 525–534.
Huang J, Wei W, Zhang J, Liu G, Bignell GR, Stratton MR et al. (2004). Whole genome DNA copy number changes identified by high density oligonucleotide arrays. Hum Genomics 1: 287–299.
Huang N, Lee I, Marcotte EM, Hurles ME (2010). Characterising and predicting haploinsufficiency in the human genome. PLoS Genet 6: e1001154.
Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y et al. (2004). Detection of large-scale variation in the human genome. Nat Genet 36: 949–951.
Ishkanian AS, Malloff CA, Watson SK, DeLeeuw RJ, Chi B, Coe BP et al. (2004). A tiling resolution DNA microarray with complete coverage of the human genome. Nat Genet 36: 299–303.
Kallioniemi A, Kallioniemi OP, Sudar D, Rutovitz D, Gray JW, Waldman F et al. (1992). Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 258: 818–821.
Kennedy GC, Matsuzaki H, Dong S, Liu WM, Huang J, Liu G et al. (2003). Large-scale genotyping of complex DNA. Nat Biotechnol 21: 1233–1237.
Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T et al. (2008). Mapping and sequencing of structural variation from eight human genomes. Nature 453: 56–64.
Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, Simons JF et al. (2007). Paired-end mapping reveals extensive structural variation in the human genome. Science 318: 420–426.
LaFramboise T (2009). Single nucleotide polymorphism arrays: a decade of biological, computational and technological advances. Nucleic Acids Res 37: 4181–4193.
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J et al. (2001). Initial sequencing and analysis of the human genome. Nature 409: 860–921.
Le Caignec C, Spits C, Sermon K, De Rycke M, Thienpont B, Debrock S et al. (2006). Single-cell chromosomal imbalances detection by array CGH. Nucleic Acids Res 34: e68.
Li H, Ruan J, Durbin R (2008). Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res 18: 1851–1858.
Lichter P, Cremer T, Borden J, Manuelidis L, Ward DC (1988). Delineation of individual human chromosomes in metaphase and interphase cells by in situ suppression hybridization using recombinant DNA libraries. Hum Genet 80: 224–234.
Linardopoulou EV, Williams EM, Fan Y, Friedman C, Young JM, Trask BJ (2005). Human subtelomeres are hot spots of interchromosomal recombination and segmental duplication. Nature 437: 94–100.
Maher CA, Kumar-Sinha C, Cao X, Kalyana-Sundaram S, Han B, Jing X et al. (2009). Transcriptome sequencing to detect gene fusions in cancer. Nature 458: 97–101.
Mani RS, Chinnaiyan AM (2010). Triggers for genomic rearrangements: insights into genomic, cellular and environmental influences. Nat Rev Genet 11: 819–829.
Mantripragada KK, Tapia-Paez I, Blennow E, Nilsson P, Wedell A, Dumanski JP (2004). DNA copy-number analysis of the 22q11 deletion-syndrome region using array-CGH with genomic and PCR-based targets. Int J Mol Med 13: 273–279.
Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K et al. (2009). Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 361: 1058–1066.
Marioni JC, Thorne NP, Valsesia A, Fitzgerald T, Redon R, Fiegler H et al. (2007). Breaking the waves: improved detection of copy number variation from microarray-based comparative genomic hybridization. Genome Biol 8: R228.
McCarroll SA, Kuruvilla FG, Korn JM, Cawley S, Nemesh J, Wysoker A et al. (2008). Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet 40: 1166–1174.
Medvedev P, Stanciu M, Brudno M (2009). Computational methods for discovering structural variation with next-generation sequencing. Nat Methods 6: S13–S20.
Mefford HC, Eichler EE (2009). Duplication hotspots, rare genomic disorders, and common disease. Curr Opin Genet Dev 19: 196–204.
Metzker ML (2010). Sequencing technologies—the next generation. Nat Rev Genet 11: 31–46.
Meyerson M, Gabriel S, Getz G (2010). Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet 11: 685–696.
Michalet X, Ekong R, Fougerousse F, Rousseaux S, Schurra C, Hornigold N et al. (1997). Dynamic molecular combing: stretching the whole human genome for high-resolution studies. Science 277: 1518–1523.
Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP et al. (2010). Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 86: 749–764.
Mills RE, Luttig CT, Larkins CE, Beauchamp A, Tsui C, Pittard WS et al. (2006). An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res 16: 1182–1190.
Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C et al. (2011). Mapping copy number variation by population-scale genome sequencing. Nature 470: 59–65.
Nuwaysir EF, Huang W, Albert TJ, Singh J, Nuwaysir K, Pitas A et al. (2002). Gene expression analysis using oligonucleotide arrays produced by maskless photolithography. Genome Res 12: 1749–1755.
Olshen AB, Venkatraman ES, Lucito R, Wigler M (2004). Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 5: 557–572.
Ou Z, Stankiewicz P, Xia Z, Breman AM, Dawson B, Wiszniewska J et al. (2011). Observation and prediction of recurrent human translocations mediated by NAHR between nonhomologous chromosomes. Genome Res 21: 33–46.
Pang AW, MacDonald JR, Pinto D, Wei J, Rafiq MA, Conrad DF et al. (2010). Towards a comprehensive structural variation map of an individual human genome. Genome Biol 11: R52.
Parameswaran P, Jalili R, Tao L, Shokralla S, Gharizadeh B, Ronaghi M et al. (2007). A pyrosequencing-tailored nucleotide barcode design unveils opportunities for large-scale sample multiplexing. Nucleic Acids Res 35: e130.
Park H, Kim JI, Ju YS, Gokcumen O, Mills RE, Kim S et al. (2010). Discovery of common Asian copy number variants using integrated high-resolution array CGH and massively parallel DNA sequencing. Nat Genet 42: 400–405.
Parra I, Windle B (1993). High resolution visual mapping of stretched DNA by fluorescent hybridization. Nat Genet 5: 17–21.
Peiffer DA, Le JM, Steemers FJ, Chang W, Jenniges T, Garcia F et al. (2006). High-resolution genomic profiling of chromosomal aberrations using Infinium whole-genome genotyping. Genome Res 16: 1136–1148.
Pinkel D, Landegent J, Collins C, Fuscoe J, Segraves R, Lucas J et al. (1988). Fluorescence in situ hybridization with human chromosome-specific libraries: detection of trisomy 21 and translocations of chromosome 4. Proc Natl Acad Sci USA 85: 9138–9142.
Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D et al. (1998). High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet 20: 207–211.
Pinto D, Darvishi K, Shi X, Rajan D, Rigler D, Fitzgerald T et al. (2011). Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nat Biotechnol 29: 512–520.
Pique-Regi R, Monso-Varona J, Ortega A, Seeger RC, Triche TJ, Asgharzadeh S (2008). Sparse representation and Bayesian detection of genome copy number alterations from microarray data. Bioinformatics 24: 309–318.
Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD et al. (2010). A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 463: 191–196.
Pollack JR, Perou CM, Alizadeh AA, Eisen MB, Pergamenschikov A, Williams CF et al. (1999). Genome-wide analysis of DNA copy-number changes using cDNA microarrays. Nat Genet 23: 41–46.
Price TS, Regan R, Mott R, Hedman A, Honey B, Daniels RJ et al. (2005). SW-ARRAY: a dynamic programming solution for the identification of copy-number changes in genomic DNA using array comparative genome hybridization data. Nucleic Acids Res 33: 3455–3464.
Przybytkowski E, Ferrario C, Basik M (2011). The use of ultra-dense array CGH analysis for the discovery of micro-copy number alterations and gene fusions in the cancer genome. BMC Med Genomics 4: 16.
Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD et al. (2006). Global variation in copy number in the human genome. Nature 444: 444–454.
Scherer SW, Lee C, Birney E, Altshuler DM, Eichler EE, Carter NP et al. (2007). Challenges and standards in integrating surveys of structural variation. Nat Genet 39: S7–15.
Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G (2002). Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 30: e57.
Schrock E, du Manoir S, Veldman T, Schoell B, Wienberg J, Ferguson-Smith MA et al. (1996). Multicolor spectral karyotyping of human chromosomes. Science 273: 494–497.
Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P et al. (2004). Large-scale copy number polymorphism in the human genome. Science 305: 525–528.
Sharp AJ, Itsara A, Cheng Z, Alkan C, Schwartz S, Eichler EE (2007). Optimal design of oligonucleotide microarrays for measurement of DNA copy-number. Hum Mol Genet 16: 2770–2779.
Sharp AJ, Locke DP, McGrath SD, Cheng Z, Bailey JA, Vallente RU et al. (2005). Segmental duplications and copy-number variation in the human genome. Am J Hum Genet 77: 78–88.
Shendure J, Porreca GJ, Reppas NB, Lin X, McCutcheon JP, Rosenbaum AM et al. (2005). Accurate multiplex polony sequencing of an evolved bacterial genome. Science 309: 1728–1732.
Simonis M, Klous P, Homminga I, Galjaard RJ, Rijkers EJ, Grosveld F et al. (2009). High-resolution identification of balanced and complex chromosomal rearrangements by 4C technology. Nat Methods 6: 837–842.
Skinner BM, Robertson LB, Tempest HG, Langley EJ, Ioannou D, Fowler KE et al. (2009). Comparative genomics in chicken and Pekin duck using FISH mapping and microarray analysis. BMC Genomics 10: 357.
Slade I, Stephens P, Douglas J, Barker K, Stebbings L, Abbaszadeh F et al. (2010). Constitutional translocation breakpoint mapping by genome-wide paired-end sequencing identifies HACE1 as a putative Wilms tumour susceptibility gene. J Med Genet 47: 342–347.
Snijders AM, Nowak N, Segraves R, Blackwood S, Brown N, Conroy J et al. (2001). Assembly of microarrays for genome-wide measurement of DNA copy number. Nat Genet 29: 263–264.
Solinas-Toldo S, Lampel S, Stilgenbauer S, Nickolenko J, Benner A, Dohner H et al. (1997). Matrix-based comparative genomic hybridization: biochips to screen for genomic imbalances. Genes Chromosomes Cancer 20: 399–407.
Speicher MR, Carter NP (2005). The new cytogenetics: blurring the boundaries with molecular biology. Nat Rev Genet 6: 782–792.
Speicher MR, Gwyn Ballard S, Ward DC (1996). Karyotyping human chromosomes by combinatorial multi-fluor FISH. Nat Genet 12: 368–375.
Stankiewicz P, Lupski JR (2010). Structural variation in the human genome and its role in disease. Annu Rev Med 61: 437–455.
Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ et al. (2011). Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144: 27–40.
Stephens PJ, McBride DJ, Lin ML, Varela I, Pleasance ED, Simpson JT et al. (2009). Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature 462: 1005–1010.
Sudmant PH, Kitzman JO, Antonacci F, Alkan C, Malig M, Tsalenko A et al. (2010). Diversity of human copy number variation and multicopy genes. Science 330: 641–646.
Talseth-Palmer BA, Bowden NA, Hill A, Meldrum C, Scott RJ (2008). Whole genome amplification and its impact on CGH array profiles. BMC Res Notes 1: 56.
Telenius H, Carter NP, Bebb CE, Nordenskjold M, Ponder BA, Tunnacliffe A (1992). Degenerate oligonucleotide-primed PCR: general amplification of target DNA by a single degenerate primer. Genomics 13: 718–725.
The International HapMap Consortium (2003). The International HapMap Project. Nature 426: 789–796.
Tuzun E, Sharp AJ, Bailey JA, Kaul R, Morrison VA, Pertz LM et al. (2005). Fine-scale structural variation of the human genome. Nat Genet 37: 727–732.
Tzschach A, Bisgaard AM, Kirchhoff M, Graul-Neumann LM, Neitzel H, Page S et al. (2010). Chromosome aberrations involving 10q22: report of three overlapping interstitial deletions and a balanced translocation disrupting C10orf11. Eur J Hum Genet 18: 291–295.
Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG et al. (2001). The sequence of the human genome. Science 291: 1304–1351.
Volker M, Backstrom N, Skinner BM, Langley EJ, Bunzey SK, Ellegren H et al. (2010). Copy number variation, chromosome rearrangement, and their association with recombination during avian evolution. Genome Res 20: 503–511.
Wiegant J, Kalle W, Mullenders L, Brookes S, Hoovers JM, Dauwerse JG et al. (1992). High-resolution in situ hybridization using DNA halo preparations. Hum Mol Genet 1: 587–591.
Winchester L, Yau C, Ragoussis J (2009). Comparing CNV detection methods for SNP arrays. Brief Funct Genomic Proteomic 8: 353–366.
Yamada NA, Rector LS, Tsang P, Carr E, Scheffer A, Sederberg MC et al. (2011). Visualization of fine-scale genomic structure by oligonucleotide-based high-resolution FISH. Cytogenet Genome Res 132: 248–254.
Yamazawa K, Ogata T, Ferguson-Smith AC (2010). Uniparental disomy and human disease: an overview. Am J Med Genet C Semin Med Genet 154C: 329–334.
Ye K, Schulz MH, Long Q, Apweiler R, Ning Z (2009). Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25: 2865–2871.
Yoon S, Xuan Z, Makarov V, Ye K, Sebat J (2009). Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res 19: 1586–1592.
Acknowledgements
We thank N Carter for commenting on the manuscript and T Fitzgerald for helpful discussions. SL and SMG are supported by the Wellcome Trust [Grant no. WT098051].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Heredity website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Le Scouarnec, S., Gribble, S. Characterising chromosome rearrangements: recent technical advances in molecular cytogenetics. Heredity 108, 75–85 (2012). https://doi.org/10.1038/hdy.2011.100
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2011.100
Keywords
This article is cited by
-
Successful pregnancy after prenatal diagnosis by NGS for a carrier of complex chromosome rearrangements
Reproductive Biology and Endocrinology (2020)
-
Next-generation sequencing approaches for the study of genome and epigenome toxicity induced by sulfur mustard
Archives of Toxicology (2018)
-
Explorations to improve the completeness of exome sequencing
BMC Medical Genomics (2016)
-
Polyploid and aneuploid detection in apple using a single nucleotide polymorphism array
Tree Genetics & Genomes (2015)
-
Study of structural chromosome abnormalities to increase the understanding of human genetic diversity: a commentary on signature of backward replication slippage at the copy number variation junction
Journal of Human Genetics (2014)
