
Abstract
How do morphological differences between species evolve at the genetic level? This study investigates the genetic basis of recent divergence in male wing size between species of the model parasitoid wasp Nasonia. The forewings of flightless Nasonia vitripennis males are 2.3 times smaller than males of their flighted sister species N. giraulti. We describe a major genetic contributor to this difference: the sex-specific widerwing (wdw) locus, which we have backcrossed from N. giraulti into N. vitripennis and mapped to an 0.9 megabase region of chromosome 1. This introgression of wdw from large-winged N. giraulti into small-winged N. vitripennis increases male but not female forewing width by 30% through wing region-specific size changes. Indirect evidence suggests that cell number changes across the wing explain the majority of the wdw wing-size difference, whereas changes in cell size are important in the center of the wing. Introgressing the same locus from the other species in the genus, N. longicornis and N. oneida, into N. vitripennis produces intermediate and large male wing sizes. To our knowledge, this is the first study to introgress a morphological quantitative trait locus (QTL) from multiple species into a common genetic background. Epistatic interactions between wdw and other QTL are also identified by introgressing wdw from N. vitripennis into N. giraulti. The main findings are (1) the changes at wdw have sex- and region-specific effects and could, therefore, be regulatory, (2) the wdw locus seems to be a co-regulator of cell size and cell number, and (3) the wdw locus has evolved different wing width effects in three species.
Similar content being viewed by others

Introduction
A primary goal of biology is to understand the mechanisms behind the diversity of phenotypes among species. Genetic investigation of morphological differences between species has emerged only recently (Orr, 2001). As morphological differences often involve the modification of basic developmental processes, careful study of these traits can also reveal developmental mechanisms of patterning, growth, and size control (Stern, 2000). For example, a major component of the size difference between wild and domesticated tomato fruit has been found to act by changing cell number (Frary et al., 2000). This gene, fw2.2, was positionally cloned and subsequently implicated as a regulator of the cell cycle in plants (Cong and Tanksley, 2006).
Recently, controversy has been building over what types of genetic alteration can cause the sex- and organ-specific changes that are seen in many cases of morphological evolution (for example, Hoekstra and Coyne, 2007; Lynch and Wagner, 2008; Stern and Orgogozo, 2008). Basically, some have argued that noncoding cis-regulatory mutations are a primary source of morphological novelty because regulatory change is thought to be more likely to have spatially or temporally restricted effects than protein sequence change (Stern, 2000; Carroll, 2005). Others argue that this conclusion is premature: a number of other types of mutations that affect gene regulation have been identified and may be important in morphological evolution (Hoekstra and Coyne, 2007; Lynch and Wagner, 2008). The controversy is perhaps most acute because relatively few morphological differences between species have been mapped to a fine enough genetic level to ascertain the genetic cause (Hoekstra and Coyne, 2007; Stern and Orgogozo, 2008).
Sexually dimorphic traits are often the most rapidly evolving traits between species, making them prime candidates for dissection of the genetic basis of development (Kopp and True, 2002). For example, investigation of a recently evolved sex-specific abdominal pigmentation difference between Drosophila species led to the identification of the bric-a-brac gene as a major contributor (Kopp et al., 2000). The pigmentation difference caused by the bric-a-brac locus involves changes in cis-regulatory binding sites for the sex signaling gene Doublesex and the abdominal patterning gene ABD-B (Williams et al., 2008). Investigation of recently evolved sexual differentiation can, therefore, lead to understanding how gene networks acquire sex and tissue specificity in the early stages of their evolution.
Morphological differences between species often involve changes at multiple genetic loci, as revealed by F2 quantitative trait locus (QTL) mapping (for example, Alpert et al., 1995; Orr, 2001; Gadau et al., 2002). A powerful approach to reducing this complexity is introgression of individual QTL through backcrossing into a common genetic background. The introgression approach creates an effectively Mendelian locus, which can be analyzed in detail, both phenotypically and genetically. Between-species introgression lines have been widely used to isolate QTL behind important morphological and physiological differences in cultivated plants and their wild relatives (Lippman et al., 2007). The tomato fw2.2 QTL mentioned above was found in a between-species introgression line, which made it straightforward for the causative gene to be positionally cloned (Frary et al., 2000). In animals, introgression lines have been used in relatively few cases to isolate morphological differences between species, for example sex comb morphology in Drosophila (Graze et al., 2007), wing size in Nasonia (Weston et al., 1999), and male fertility traits in mice (L’Hôte et al., 2007).
The insect wing is essentially a two-dimensional sheet of cells and is, therefore, a relatively simple model in which to study the cellular mechanisms behind morphological evolution. Specifically, changes in the size and shape of the wing can be readily attributed to changes in size, shape, and number of cells (Dobzhansky, 1929). Studies of wing size and shape variation in Drosophila melanogaster have revealed, among other things, that cell size and cell number may be under compensatory regulation (McCabe et al., 1997). A number of genes, which, when mutated, affect wing shape and cell size have been identified. These are often members of conserved growth regulation pathways such as cell cycle, insulin signaling, and apoptosis (Hafen and Stocker, 2003). Studies of quantitative wing shape variation within D. melanogaster have implicated several of these genes as potential contributors to intraspecific variation (Dworkin and Gibson, 2006; Weber et al., 2008). Wing shape across the Drosophila genus is remarkably conserved (Houle et al., 2003) and may be under stabilizing selection or canalization (Gilchrist et al., 2000).
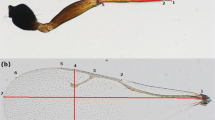
To make full use of the wing as a model of the cellular and genetic basis of morphological evolution, it will be necessary to also study larger differences in wing size and shape. Here, we investigate the genetics of recent sex-specific wing-size evolution in Nasonia, a genus of parasitic wasps containing four species, which have dramatic differences in male wing size (Figure 1; Darling and Werren, 1990; Raychoudhury et al., 2009, 2010). Nasonia is an emerging genetic model of the evolution of species differences: the species are interfertile after removal of Wolbachia bacteria, allowing introgression of QTL for wing size and other traits between species (Breeuwer and Werren, 1995; Weston et al., 1999; Clark et al., 2010; Desjardins et al., 2010). In addition, three species’ genomes have now been sequenced and a range of genomic and genetic resources have been produced (Werren et al., 2010). It should also be emphasized that Nasonia males are haploid, which simplifies the genetic analysis of complex traits (Weston et al., 1999; Werren and Loehlin, 2009).

Male forewing size differs across the Nasonia genus and in introgressions of the wdw locus from each species, but female forewings of wdw introgressions are not affected. Cladogram depicts relationships between the Nasonia species (Raychoudhury et al., 2009). Scale bar: 200 μm.
The most striking morphological difference between Nasonia species is the 2.3-fold male-specific wing-size difference between small-winged Nasonia vitripennis and large-winged N. giraulti (Darling and Werren, 1990; Weston et al., 1999). QTL underlying this difference have been mapped (Gadau et al., 2002), as have QTL for wing length differences between N. vitripennis and medium-winged N. longicornis (Rütten et al., 2004). These analyses have revealed both large effect and epistatic contributors, albeit with a limited level of resolution. Using an introgression approach, one large-effect wing-size locus (ws1) that increases male wing size by 63% has been isolated from N. giraulti by introgression into N. vitripennis (Weston et al., 1999). We have subsequently been able to use ws1 introgression lines to determine that cell size and cell number changes contribute about equally to its wing-size change and to identify the genetic basis of ws1 by positional cloning (Loehlin et al., 2010).
Here, we report the discovery of a second large-effect wing-size QTL in Nasonia, called widerwing (wdw). We identify its genomic location, sex specificity, and morphological effects in multiple introgression lines. Introgression of this QTL from N. giraulti into an inbred N. vitripennis background increases the wing width in males but not females. An 880 kilobase region containing wdw is identified from the overlap of three introgression lines, leading the way to future positional cloning. The effects of wdw on cell size and cell number in different regions of the wing are inferred using the pattern of seta on the wing surface. Introgression lines from two other Nasonia species reveal that the wdw QTL has diverged in multiple lineages. Epistatic interaction of wdw with other QTL is shown using a reciprocal introgression of N. vitripennis wdw into N. giraulti. These approaches allow us to characterize the wdw QTL to a level of detail not possible in standard F2 QTL analyses.
Materials and methods
Widerwing introgressions
To identify the genetic basis of male-specific wing size, we conducted a set of experiments to backcross male-specific wing QTL from N. giraulti into N. vitripennis. Here, we describe the three introgression strains (B3, D1, and SB2C) that contain the wdw QTL. Other wing-size QTL from this screen will be described elsewhere. The B3 and D1 strains were isolated from an experiment in which N. giraulti males from wild-type strain RV2R were first mated to N. vitripennis females from visible marker strain bl13,st5219. F1 females were then backcrossed to bl13,st5219 males and F2 sublines were founded. Subsequent backcrossing of hybrid females was to males of the wild-type N. vitripennis LabII strain, removing the unlinked visible markers in the process. Each generation of the backcross, hybrid virgin females were given two Sarcophaga bullata hosts to produce haploid male offspring for 48 h. The females were then mated to LabII males and given hosts to produce female offspring. We examined the wing size of male offspring of the virgin females and selected females who were segregating for apparently Mendelian male wing-size loci. Virgin females for the next generation of the backcross were collected from the offspring of these selected females (as in Weston et al., 1999). After 8–12 generations of backcrossing, strains carrying single wing-size QTL were made homozygous. The SB2C strain was isolated similarly, except that the initial cross was between different strains: N. vitripennis strain st5219,or123 females and N. giraulti strain R16A males (RV2R and R16A are both descendents of the same inbred laboratory strain, RV2, but with different mitochondria; Breeuwer and Werren, 1995). Subsequently, B3, D1, and SB2C were found to contain the same QTL, termed as widerwing (wdw). The B3 strain was later cured of its Wolbachia infection and backcrossed into the N. vitripennis strain used for genome sequencing (AsymCX) for 14 generations, to produce a homogenous genetic background. The resulting strain (B3bc14) was used for subsequent analyses and is referred to in this paper as wdwgV (that is, wdw from N. giraulti in N. vitripennis background).
The N. longicornis allele of wdw was similarly isolated, starting with males of the genome-sequenced IV7u strain and backcrossing into N. vitripennis (AsymCX) for eight generations. A linked molecular marker discovered in the mapping experiments (S1-i534) was used to select for heterozygous females. Two strains were derived independently after the F1 generation, wdwlV-A and wdwlV-B, and made homozygous. One N. oneida wdw introgression into N. vitripennis, wdwoV-B, was isolated from males of an antibiotically treated N. oneida female (NoNYBr1136 strain) using the same procedure. Similarly, wdw was backcrossed from N. vitripennis (AsymCX) into N. giraulti (R16A) in nine generations, producing the wdwvG-A introgression strain.
Visible marker mapping
Initial mapping suggested distant linkage between wdw and the Linkage Group II (=chromosome 1, Rütten et al., 2004) marker reddish5 (rdh5). Visible mapping was, therefore, conducted between wdwgV, rdh5 and a third chromosome 1 marker, distantennapedia (dant) (Werren and Perrot-Minnot, 1999). The dant males were mated to B3,rdh5 females and then F1 females were hosted as virgins to generate a visible mapping population of haploid F2 males.
Genotyping
Polymerase chain reaction genotyping assays were developed using the Nasonia vitripennis 1.0 genome assembly and N. giraulti and N. longicornis Sanger sequencing reads, which were aligned with NCBI BLAST and Sequencher (Gene Codes). We designed primers to amplify species-specific length polymorphisms (indels) identified on genome assembly SCAFFOLD1. Sequences for these wdw-region primers are:
S1-i451-F CTGTTGTTGTACGCCCTGATTCC, S1-i451-R AACCAGAGTCCTAAAGCCAGCC, Nv: 563 bp, Ng: 501 bp. S1-i481-F2 TTTTGGCGGGAAATCTCG, S1-i481-R2 GGCTGACTTATGGGCGATTCTC, Nv: 781 bp, Ng: 719 bp. S1-i534-F TCCAACCATTTGTTTATGTAATACCG, S1-i534-R GCAGATTCATCCGAAATAAGCG, Nv: 486 bp, Ng: 527 bp. S1-i540-F GTGGCGTTTGGCGGTTAC, S1-i540-R GTTTTTCAAATACGCACACTTCC, Nv: 455 bp, Ng: 535 bp. Genomic locations are given in Appendix 1 (see Supplementary data). Each of these assays was found to discriminate N. vitripennis alleles from N. giraulti, N. oneida, and N. longicornis.
Genomic DNA was extracted from single wasps using the Squish protocol (Gloor and Engels, 1992). All polymerase chain reaction reactions were carried out using 14.3 μl ddH2O, 2 μl 10 × polymerase chain reaction buffer, 0.6 μl 10 mM MgCl, 0.4 μl 10 mM dNTPs, 0.8 μl 10 mM forward primer, 0.8 μl 10 mM reverse primer, 0.1 μl 5 μ μl–1 Taq Polymerase (Invitrogen, Carlsbad, CA, USA), and 1 μl genomic DNA (males) or 0.2 μl genomic DNA (females). Reaction cycle for all primer pairs in iCycler (Bio-Rad, Hercules, CA, USA) thermalcyclers was 94C for 2 min, 36 repeats of (94 °C for 30 s–55 °C for 30 s–72 °C for 60 s), and 72 °C for 10 min.
Wing measurements
For wing measurements, individual females were provided two fly hosts for 48 h in a vial at 25 °C, and measurements were made on the offspring. This design reduces allometric effects of crowding on body size because fewer offspring are produced than can consume the host (Weston et al., 1999). Wild-type strains used were AsymC (N. vitripennis), R16A (N. giraulti), IV7u (N. longicornis), and NoNYBr1136 (N. oneida). Male wing samples were collected from the offspring of virgin females, whereas female wing samples were collected from the offspring of mated females. The one exception to this was wdwvG. Male samples of this strain were collected from the offspring of cultures containing five mated females and five hosts, as single-female settings only produced diapause larvae. Wings from 5 offspring per vial and 4–10 vials per genotype were collected and mounted (Table 1); occasionally, damaged mounts meant that fewer than five wings were measured per vial. Adult fore- and hindwings were dissected at the hinge adjoining the thorax and dry mounted on microscope slides under coverslips. Heads were mounted on double-sided tape to provide a comparative measure of body size. Wings were photographed on a Zeiss AxioImager Z1 compound scope at × 10 as mosaic images. For wing seta counts, wings were re-imaged at × 20 through multiple focal planes (z-stacks). Heads were imaged at × 4 under darkfield and green autofluorescence to highlight the eyes.
Measurements were performed on the wing images using AxioVision 4.6 software (Carl Zeiss, Thornwood, NY, USA). In this study, wing length is defined as the maximum distance between the notch at the proximal edge of the costal cell and the distal part of the wing (Figure 2). Wing width is the perpendicular distance between two lines parallel to the length axis that intersect the most anterior and posterior points of the wing. These length and width measurements also defined the proximal-distal (P-D) axis and the anterior–posterior (A-P) axis, respectively (Figure 2). Wing area was measured by drawing an outline around the wing, starting at the notch. Head width (interocular width), a measure of body size, was measured as the length of a line segment with end points at the inner edges of the compound eyes that is tangent to the anterior edges of the paired ocelli.

Landmarks, axes, and wing regions used in this study. Black lines represent the length (P-D) and width (A-P) axes. Arrowheads show forewing landmarks used for calculating distances along the A-P axis. Regions used for seta counts are depicted with numbers and are defined by landmarks (white circles) placed at the intersection of either the wing margin, folds (thick white curves) or translations of the A-P and P-D axes (thin white lines). (a) Landmarks on a wdwgV forewing. (b) Landmarks on a wdwvV (N. vitripennis) forewing.
Linear measurements of forewing sections were calculated from the position of landmarks placed along the P-D and A-P axes. Forewing landmark positions for this series of measurements are shown in Figure 2 (arrowheads). To determine forewing landmarks along the A-P (width) axis, a line parallel to the A-P axis was drawn tangent to the distal part of the stigma. Landmarks were then placed at the intersections between the line and the various veins and folds (Figure 2).
Regional seta size, number, and interseta shape measurements
Prominent, hairlike setae are produced by cells on the wing blade and have been used in other studies (particularly in Drosophila) to infer cell size and cell number contributions to wing size (for example, Zwaan et al., 2000). Local differences in cell size can be inferred on the basis of changes in area per seta, whereas cell number differences can be inferred on the basis of changes in seta number. We have found a 3:1 ratio of cells per seta in both N. vitripennis and another introgressed wing-size QTL (ws1) despite large differences in cell size and number between the two (Loehlin et al., 2010). However, we have not experimentally determined the relationship between seta number and cell number for the wdw QTL. Therefore, we refer to changes in seta number, area per seta, and interseta shape specifically rather than the inferred mechanisms of cell number, area, and shape change.
Locations of all setae on the dorsal forewing surface were determined for a subset of wings (one male wing per vial). Coordinates of all dorsal setae were recorded from × 20 z-stacked wing images, excluding setae at the wing margin and on the darkened vein. For interseta shape measurements, each seta's nearest neighbors in the anterior, posterior, proximal, and distal directions were identified using a Perl script. Interseta shape was calculated as the ratio of A-P to P-D nearest neighbor distances. Wings were then divided into 12 regions using the landmarks shown in Figure 2. Region area was calculated from landmark coordinates or manually outlined (regions 5 and 9) to exclude the stigma and include the wing margin curvature. Each seta was assigned to a region and the regional seta number and average interseta shape were computed. Area per seta for each region was calculated by dividing the region area by the number of setae.
Statistics
Pairwise comparisons between strains were conducted using Tukey's Honestly Significant Difference (HSD), on the basis of ANOVAs with vial as a nesting factor. Sample size (number of vials) is denoted by b (Table 1). As several morphological variables were measured per wing, we used the conservative Bonferroni correction for multiple tests. P-values shown were corrected by multiplying by the number of tests conducted in each analysis (basic wing measurements (Table 1), 7 variables; linear wing sections (Table 2), 8 variables; regional seta patterns (Figure 4), 52 variables).
We tested for compositional epistasis between wdw and the genetic (species) background using an additive and a multiplicative model of wing length and width. In the additive null model, if there is no epistasis, there should be no difference between (wdwgV−wdwvV) and (wdwgG−wdwvG). In the multiplicative null model, there should be no difference between (wdwgV/wdwvV) and (wdwgG/wdwvG). T-tests (planned comparisons on the basis of nested ANOVAs) were calculated for the additive and multiplicative models using variances for the difference and ratio of two independent random variables, respectively.
Results
Isolation of widerwing introgression lines
Major-effect wing-size QTL were backcrossed from large-winged N. giraulti into small-winged N. vitripennis. Three Mendelian segregating wing width QTL were isolated and made into homozygous introgression strains (B3, D1, and SB2C). All F2 male offspring of crosses between the three strains had the same wider wing phenotype, indicating that the three strains contained the same QTL. This locus was named widerwing (wdw) for its distinct width effect. This wdwgV introgression phenotype can be readily distinguished by eye from the ‘tapered’ shape wings of N. vitripennis (wdwvV) males. In particular, wdwgV gives the forewing a specific ‘triangular’ shape and greatly broadens the spacing between the stigmal vein and the median fold, whose curvature is also changed (Figure 2). A descendant of the B3 strain, called B3bc14, was chosen for detailed analysis of wing features because it is in the same background used to produce wdw introgressions from the other species. Forewing length, width, and area are not significantly different between males of the B3, D1, SB2C, or B3bc14 strains (Tukey's HSD, P<0.05; n=[40,20,20 and 40] males from b=[8,4,4 and 9] vials).
Mapping the widerwing introgressions
A future goal is to identify the wdw gene by positional cloning. As a first step, we identified the location of N. giraulti DNA in the wdwgV lines using the newly sequenced Nasonia genomes. We first mapped the wdwgV introgression phenotype using visible markers. The wdw phenotype shows linkage to the Linkage Group II (Chromosome 1, Rütten et al., 2004) markers rdh5 and dant (χ2 tests, P<0.01, n=696). Map distances are 61 cM (Kosambi corrected) between wdw and rdh5, 78 cM between wdw and dant, and 21 cM between rdh5 and dant.
We used this visible map to estimate the location of the wdwgV introgressions in the sequenced N. vitripennis and N. giraulti genomes. A molecular marker in the inferred wdw region (NVC1_13 in SCAFFOLD1, Niehuis et al., 2008) was identified on the basis of its map position relative to a homolog of the Drosophila gene spineless-aristapedia, which was proposed to be a candidate for the dant mutation (Werren and Perrot-Minnot, 1999). The NVC1_13 marker shows giraulti genotypes for two of the three wdwgV introgressions (Appendix 1 in Supplementary data). Additional markers were then designed along SCAFFOLD1 and used to define the introgressed giraulti region within the three wdwgV lines (Figure 3; Appendix 1 in Supplementary data). Confirming that this was the correct region, the wdw phenotype cosegregated perfectly with the i481 and i540 markers in 180 F2 male offspring of crosses between the D1 introgression and wild-type N. vitripennis. The shared overlapping giraulti regions of the three wdwgV lines define the possible location of wdw to a region of 880 kilobases (Figure 3).

Map of the wdw region. Genotype at length polymorphism (indel) markers is shown for the three wdwgV introgressions. Dotted line: Consensus location of the wdw QTL on the basis of shared overlapping markers in the region. Black bars: N. vitripennis (background) genotype. Open bars: N. giraulti (introgression) genotype. Shading: Genotype between two markers is unknown. Marker locations in SCAFFOLD1 and map data for other introgressions are given in Appendix 1 (see Supplementary data).
Wing shape effects of the wdwgV introgression
Widerwing is a major contributor to the 2.3-fold male wing-size difference between N. vitripennis and N. giraulti (Figure 1; Table 1). The wdwgV introgression recovers 36% of the difference in forewing width and 28% of the difference in length between the species. Relative to wild-type N. vitripennis (wdwvV), the wdwgV introgression increases male forewing length by 8%, forewing width by 29%, and forewing area by 32% (Tukey's HSD, P<0.05, b=[8,9]) (Table 1). Male hindwing size is also increased, though not as severely; length and width increase by 10% and area increases by 20%. In contrast, female forewing and hindwing length, width, and area are completely unaffected by the introgression (Tukey's HSD, P>0.05, b=[8,5]). It is noteworthy that such a large-effect locus is entirely sex specific (Figure 1).
To determine whether the observed male wing shape changes derive from uniform changes or localized changes in size and shape, we divided forewings into sections using landmarks along the length (A-P) and width (P-D) axes (Figure 2a). Section distances were significantly greater across most of the wdwgV forewing when compared with wdwvV (Table 2). Large localized changes are also seen. The central section increases in width by a striking 74%, corresponding to the displacement of the median fold (Figure 2), whereas the other sections along the A-P axis increase by 16–25% (Tukey's HSD, P<0.05, b=[8,9]). Along the P-D axis, the two distal sections are 15–16% longer. The proximal section is also significantly increased, but only by 4%. To summarize, the wdwg introgression increases wing size across the forewing, but the magnitudes of these increases are region specific.
Changes to cell size, number, and shape inferred from wing seta pattern
The locally different changes in wing shape seen in the wdwgV introgression could be caused by changes in cell number, size, or shape. Setae (cell hairs) cover most of the forewing and can be used as a proxy for cells. The forewing was divided into regions on the basis of sections of the A-P and P-D axes (Figure 2) and the pattern of seta number, area per seta, and shape was determined in a subset of wings.
wdwgV increases seta number across most of the wing and area per seta at a local level (Figure 4). Seta number is significantly greater across the central and distal sections of the wdwgV forewing (Figure 4b and data not shown) (Tukey's HSD, P<0.05, n=b=8). This corresponds to a significant 43% increase in seta number across the wing (Figure 4a), suggesting an overall increase in cell number. The most dramatic local changes occur in the central-stigmal region (region 6; Figure 4b). In this region, area increases by 161%, seta number by 99%, and area per seta by 32%. Significant area per seta increases are found in this region, but not overall, suggesting a local increase in cell size. Interseta shape is significantly wider overall, but by a relatively minor 1.7%. We can, therefore, infer that wdwgV causes increases in cell number in most of the wing and increases in cell size restricted to the center of the wing.

Seta number, area, and shape contributions to forewing regions. (a) Change averaged across all 12 seta-containing regions. (b) The largest changed region (region 6) shows local area-per-seta effects. Interseta shape values greater than one indicate that seta are farther apart along the A-P (width) axis. Relative mean±s.d. is shown. Letters denote contrast groups for multiple comparisons (Tukey's HSD, n=b=8).
Widerwing from N. longicornis
We subsequently backcrossed the region containing wdw from other Nasonia species. N. longicornis and N. giraulti share a common ancestor with N. vitripennis, but are more closely related to one another (Campbell et al., 1993; Raychoudhury et al., 2009). In addition, N. longicornis male wings are intermediate in size between N. vitripennis and N. giraulti (Figure 1; Table 1). We, therefore, speculated that the wdw phenotype might be shared between N. longicornis and N. giraulti. The N. longicornis allele of wdw was backcrossed into N. vitripennis for eight generations. Molecular markers found in the wdwgV introgressions were used to select on the same genomic region during the backcrosses. Two strains were isolated: wdwlV-A and wdwlV-B. Both strains share longicornis genotypes at markers in the wdw region and most flanking regions (Appendix 1). Male wings of these two strains were not significantly different for forewing or hindwing length or width (Tukey's HSD, P>0.05, n=20, b=5), so the measurements were pooled.
The N. longicornis wdw locus (wdwlV) has an intermediate effect when introgressed into N. vitripennis (Figure 1; Table 1). Specifically, wdwlV male forewings are 11% wider and have 11% larger area than wdwvV (Tukey's HSD, P<0.05, b=[8,10]). Forewing length and hindwing length, width, and area are not significantly different (P>0.05). These changes are not as large as those seen in wdwgV; wdwlV has only 24% of the increase in forewing width and 38% of the increase in forewing area seen in wdwgV. The wdwlV introgression recovers 23% of the species difference in male forewing width between N. longicornis and N. vitripennis, which is less than the 36% recovery seen in wdwgV.
The wdwlV introgression also shows small but significant reductions in female wing size (4–6% smaller than wdwvV, Table 1; Tukey's HSD, P<0.05, b=[8,5]) and head width (4% smaller). Head width has been shown to covary with wing size (Weston et al., 1999). Including head width as a covariate with wing length and width (not shown) reduced the mean forewing length and width differences between wdwlV and wdwvV females to 2.8%, although these adjusted means were still significantly different. Male and female wdwlV wings are both smaller than wdwgV. However, the relative wing width difference is much greater in the males (area is 11% larger than wdwvV and 16% smaller than wdwgV). Further reduction of longicornis flanking regions around wdwl will be needed to resolve whether these effects are due to wdwl or linked genes.
Changes in wing sections and seta pattern in wdwlV
At both a broad and a local level, the N. longicornis introgression of wdw has wing-effects intermediate between the N. giraulti and N. vitripennis loci. The central forewing is also the largest changed section in wdwlV, though the magnitude (28%) is less than is seen in wdwgV (Table 2; Tukey's HSD, P<0.05, b=[9,10]). In contrast, the distal section of the A-P axis, which was longer in wdwgV than in wdwvV, is not significantly longer in wdwlV. This expansion of distal section length in wdwgV but not wdwlV is noteworthy, as it differs from the pattern of intermediate size seen in other wing sections and may, therefore, be a discrete effect.
The seta pattern in wdwlV is consistent with its intermediate wing size (Figure 4). In the center of the wing (region 6; Figure 4b), region area is increased by 50%, seta number is increased by 21%, and area per seta is 26% larger (Tukey's HSD, P<0.05, n=b=8). Although the area per seta increase in the center of the wing is similar to wdwgV, the seta number expansion in wdwlV seems to be more spatially restricted than with wdwgV as no seta number difference across the wing was detected (Figure 4a). Therefore, a more localized change in cell size and number may be responsible for the intermediate wing width increase in wdwlV.
Widerwing from N. oneida
N. oneida, a newly discovered species that is sister to N. giraulti, has large male wings, similar to but slightly smaller than N. giraulti (Raychoudhury et al., 2010). Backcross of the wdw region from N. oneida into N. vitripennis yielded a strain (wdwoV-B) with male forewings visibly similar to wdwgV (Figure 1). Supporting visual observations, forewing length, width, area, and hindwing length are not significantly different (Tukey's HSD, P>0.05, b=[9,9]) (Table 1). However, hindwing width is 4% smaller relative to wdwgV males (P<0.05). These effects are also male specific: females of the wdwoV-B introgression do not differ from wdwgV in forewing or hindwing length or width (Tukey's HSD, P>0.05, b=[5,5]). With the exception of male hindwing width, the wdw introgression from N. oneida has essentially the same phenotype as the introgression from its sister species N. giraulti.
N. vitripennis widerwing in the N. giraulti background
To determine whether wdw has epistatic interactions with other QTL, we performed the reciprocal introgression of the N. vitripennis allele of wdw into large-winged N. giraulti, again using a linked molecular marker. Consistent with this wdwvG strain containing the wdw QTL, forewing width is 9% smaller and hindwing width is 8% smaller than N. giraulti (wdwgG) (Tukey's HSD, P<0.05, b=[8,4]) (Table 1). Forewing length and hindwing length are not significantly different between strains (P>0.05). The most obvious similarity between the reciprocal wdwvG and wdwgV introgressions is that the giraulti alleles have wider wings in both backgrounds (Figure 5). The wdwvG introgression recovers 17% of the species difference in wing width, about half of the recovery caused by the wdwgV introgression.

Compositional epistasis between wdw and genetic background. If wdw does not interact with other QTL in the species background and wing length (for example) is additive, Δlength[V] should equal Δlength[G]. (a) Forewing length. (b) Forewing width. (c) Forewing area.
If wdw had no epistatic interactions with other QTL that differ between the species, the effects of wdw genotype and genetic background should add (or possibly multiply) together (Phillips, 2008). However, the interaction between wdw and species background is not consistent with either an additive or multiplicative model of forewing length, width, or area (t-tests, P<0.05, b=[9,8,8,4]). This can be seen visually in Figure 5, in which the distance between the smaller wings (vitripennis background) is greater than the distance between the larger wings (giraulti background). Therefore, wdw interacts epistatically with other QTL that differ between vitripennis and giraulti. Such compositionally epistatic QTL are likely to be in the same pathway as wdw and will be interesting candidates for future study. The introgression experiments do not rule out that mitochondrial type contributes to the wing phenotype, although it would be surprising that this would be sex specific. Furthermore, this would be an epistatic interaction, albeit between nuclear and mitochondrial genotypes. Epistatic interactions could also involve maternal genotype influences on male phenotype. However, our genetic analysis clearly shows that wdw is inherited in a mendelian manner from heterozygous mothers. Therefore, any maternal effects of wdw or epistatic interactors are likely to be subtle, if they occur at all.
Discussion
Widerwing, a large-effect wing-size QTL, identified and mapped by introgression
The four Nasonia species differ substantially in male wing size. We took advantage of the interspecies genetic tools in this system to introgress and characterize alleles of a large-effect QTL, wdw. The introgression approach allowed us to map the genomic location of the locus and to determine the morphological effects of microevolution at this QTL in four separate species. Differences between multiple species at a single QTL have been examined before at the level of F2 analysis (for example Alpert et al., 1995). To our knowledge, though, this is the first report of a morphological QTL introgressed from multiple species into a common genetic background.
Further highlighting the value of the introgression approach, this substantial wing width effect was not identified in the original F2 wing QTL maps (Gadau et al., 2002; Rütten et al., 2004). This is particularly remarkable because wdw introgressions recover 36% of the N. vitripennis—N. giraulti species difference in wing width in the N. vitripennis background and 17% in the N. giraulti background. A likely explanation for the absence of wdw from the F2 QTL map is the effects of a hybrid inviability locus, which has been identified 20–30 cM away from the wdw region (Niehuis et al., 2008). Such hybrid incompatibility loci limit the effectiveness of F2 mapping between species by creating ‘pseudo-linkage’ (Weston et al., 1999). However, incompatibilities can be beneficial in the introgression approach, as they select for recombinants between the incompatibility and the QTL and can, therefore, increase the effective recombination rate during the backcross.
Sex and region specificity of wdw suggests regulatory evolution
In addition to mapping the locus, a finding of this study is that the degree of male-specific wing-size change at the wdw locus parallels the overall species differences in male wing size (that is, N. giraulti large, N. longicornis intermediate, and N. vitripennis small). Sexually dimorphic traits in other taxa tend to be rapidly evolving and are, therefore, useful for investigating the evolution of development between closely related species (Kopp and True, 2002). Changes in cis-regulatory binding sites for members of the sex-determination system have been implicated in recently evolved sexual dimorphism in Drosophila (Williams et al., 2008) and could be involved here, as well.
The sex specificity and regional effects of wdw introgressions are evidence that the underlying genetic changes have low pleiotropy. Reduced pleiotropic effects are considered to be indicative of cis-regulatory rather than coding sequence changes (Carroll, 2005). Under this hypothesis, the observed low pleiotropy, sex-specific, and region-specific differences between wdw alleles indicate cis-regulatory mutations at the locus. The generality of the cis-regulatory hypothesis is controversial, however. Existing evidence for cis-regulatory causes of morphological evolution is limited and cis-regulation is not the only possible molecular change through which pleiotropy can be reduced (Hoekstra and Coyne, 2007; Lynch and Wagner, 2008). Currently, we do not know whether cis-regulatory or coding changes are involved at wdw, and further fine scale mapping and functional analysis are needed to resolve this question.
Multiple genetic changes at the wdw locus occurred across species
The wdw loci from several Nasonia species have varying effects on wing length and width in the common N. vitripennis background. What is surprising about this result is that the same locus is responsible for multiple cases of wing-size evolution within the relatively short divergence time of the Nasonia genus (<1 MYA, Cambpell et al., 1993). This raises the question of whether the phenotypic differences between wdwvV, wdwlV, and wdwgV are due to multiple changes in a single gene, or at different, but tightly linked genes. At the present megabase resolution of mapping, we cannot distinguish between these possibilities. A case of multiple substitutions at the same morphological locus (cis-regulatory elements of the shavenbaby gene), each with isolated regional effects, has been reported for larval trichome patterns between Drosophila species (McGregor et al., 2007). We similarly observed a combination of discrete and continuous lineage-specific wing region effects in the wdw introgressions from three species. The wdw locus could, therefore, represent another case of multiple substitutions affecting the same gene. Involvement of a single locus in evolution of sex-specific wing-size regulation in three different species would imply that only a few targets are available for such evolutionary changes.
wdw interacts with other diverged QTL
Reciprocal introgression showed that the effects of wdwg and wdwv differ on the basis of the genetic (species) background. Such epistatic effects are evidence of genetic interaction, and in this case, epistasis reveals that genes that interact with wdw have also diverged between N. vitripennis and N. giraulti. Identifying the location and genetic basis of these QTL has the potential to uncover multiple components of the sex-specific wing-size gene network in Nasonia.
Cell size, cell number, and organ-size evolution
How the size of organs such as the wing is established has long interested biologists (Dobzhansky, 1929; Leevers and McNeill, 2005). The size of an organ is determined ultimately by the size and number of its cells. In recent years, a number of genes involved in regulation of cell size and number have been identified by mutation analysis. These include members of the Insulin Receptor and Tor signaling pathways (cell size and number regulation) and the cell cycle and Hippo signaling pathways (cell proliferation and apoptosis) (Johnston and Gallant, 2002; Leevers and McNeill, 2005). These pathways may not be directly involved in the regulation of organ size, however, because other signals seem to compensate organ size by increasing cell size when cell number is experimentally reduced and vice versa (Crickmore and Mann, 2008). Organ size may instead be established independently of cell size and number by instructive patterning signals such as secreted morphogens or cell adhesion genes, which have been proposed to potentiate physical-size sensing between cells (Leevers and McNeill, 2005; Crickmore and Mann, 2008). Although much is known about how these pathways operate in Drosophila and mice, which pathways are important for establishing differences in organ size remains unexplored. We are aware of only one gene that has been identified as causing the evolution of organ size: fw2.2, a previously undescribed cell cycle regulator in plants (Cong and Tanksley, 2006). Identifying the mechanisms behind more cases of cell size and number evolution should lead to further understanding of how organ size is determined.
We found that a combination of broad seta number and localized seta area changes contribute to interspecific wing-size differences at the wdw locus. In an investigation of clinal variation in Drosophila wing size, Zwaan et al. (2000) found that cell number (as estimated by setae) was the predominant mechanism behind variation in wing area as well, and concluded that cell size and cell number compensate one another in that system. With wdw, we see no evidence of compensation between cell size and number. This result suggests either that wdw operates at the level of patterning or else that the organ-size compensation mechanism may not be as important as has been argued.
The wdw QTL has two effects: a region-specific difference in area per seta (cell size) and a broader change to seta number (cell number). It is possible that multiple genes in the wdw region separately encode the cell size and cell number effects. Assuming that this is not the case, how could changes to one gene increase cell size in one region, while simultaneously increasing cell number across the wing? One hypothesis is a change in morphogen pattern. Specifically, wing cell lineages in certain regions might respond differently to changes in a single wdw signal. Second, changes in sensitivity to the concentration gradient of a morphogen could give a similar result. Third, the regional area per seta change could be an indirect effect of increasing wing width, such as by diluting an inhibitory effect of the wing vein on cell size.
Future directions
This study raises a number of questions about the structure of the wdw locus, whether the underlying changes are cis-regulatory, and how wdw regulates cell size and cell number. These can only be answered by positional cloning the wdw QTL and by identifying the genes downstream of wdw. Positional cloning and gene expression studies are practical in Nasonia, given the advantages of male haploidy and a suite of genome resources (Loehlin et al., 2010; Werren et al., 2010). Interspecies introgression is a powerful genetic tool. Our finding that introgression is practical between four different Nasonia species opens the door to future studies of a wide range of complex traits.
Conflict of interest
The authors declare no conflict of interest.
References
Alpert KB, Grandillo S, Tanksley SD (1995). fw2.2: a major QTL controlling fruit weight is common to both red- and green-fruited tomato species. Theor Appl Genet 91: 994–1000.
Breeuwer JAJ, Werren JH (1995). Hybrid breakdown between two haplodiploid species: the role of nuclear and cytoplasmic genes. Evolution 49: 705–717.
Campbell BC, Steffen-Campbell JD, Werren JH (1993). Phylogeny of the Nasonia species complex (Hymenoptera: Pteromalidae) inferred from an internal transcribed spacer (ITS2) and 28S rDNA sequences. Insect Mol Biol 2: 225–237.
Carroll SB (2005). Evolution at two levels: on genes and form. PLoS Biol 3: 1159–1166.
Clark ME, O'Hara FP, Chawla A, Werren JH (2010). Behavioural and spermatogenic hybrid male breakdown in Nasonia. Heredity (e-pub ahead of print 20 January 2010; doi:10.1038/hdy.2009.152).
Cong B, Tanksley SD (2006). FW2.2 and cell cycle control in developing tomato fruit: a possible example of gene co-option in the evolution of a novel organ. Plant Mol Biol 62: 867–880.
Crickmore MA, Mann RS (2008). The control of size in animals: insights from selector genes. BioEssays 30: 843–853.
Darling DC, Werren JH (1990). Biosystematics of Nasonia (Hymenoptera: Pteromalidae): two new species reared from birds’ nests in North America. Ann Entomol Soc Am 83: 352–370.
Desjardins CA, Perfectti F, Bartos JD, Enders LS, Werren JH (2010). The genetic basis of interspecies host preference differences in the model parasitoid Nasonia. Heredity (e-pub ahead of print 20 January 2010; doi:10.1038/hdy.145).
Dobzhansky TH (1929). The influence of the quantity and quality of chromosomal material on the size of the cells in Drosophila melanogaster. Wilhelm Roux Arch Entwickl Mech Org 115: 363–379.
Dworkin I, Gibson G (2006). Epidermal growth factor receptor and transforming growth factor-ß signaling contributes to variation for wing shape in Drosophila melanogaster. Genetics 173: 1417–1431.
Frary A, Nesbitt CT, Frary A, Grandillo S, van der Knaap E, Cong B et al. (2000). Fw2.2: a quantitative trait locus key to the evolution of tomato fruit size. Science 289: 85–88.
Gadau J, Page RE, Werren JH (2002). The genetic basis of the interspecific differences in wing size in Nasonia (Hymenoptera: Pteromalidae): major quantitative trait loci and epistasis. Genetics 161: 673–684.
Gilchrist AS, Azevedo RBR, Partridge L, O’Higgins P (2000). Adaptation and constraint in the evolution of Drosophila melanogaster wing shape. Evol Dev 2: 114–124.
Gloor GB, Engels WR (1992). Single-fly DNA preps for PCR. Drosophila Inf Serv 71: 148–149.
Graze RM, Barmina O, Tufts D, Naderi E, Harmon KL, Persianinova M et al. (2007). New candidate genes for sex-comb divergence between Drosophila mauritiana and Drosophila simulans. Genetics 176: 2561–2576.
Hafen E, Stocker H (2003). How are the sizes of cells, organs and bodies controlled? PloS Biol 1: 319–323.
Hoekstra HE, Coyne JA (2007). The locus of evolution: evo devo and the genetics of adaptation. Evolution 61: 995–1016.
Houle D, Mezey J, Galpern P, Carter A (2003). Automated measurement of Drosophila wings. BMC Evol Biol 3: 25.
Johnston LA, Gallant P (2002). Control of growth and organ size in Drosophila. BioEssays 24: 54–64.
Kopp A, True JR (2002). Evolution of male sexual characters in the oriental Drosophila melanogaster species group. Evol Dev 4: 278–291.
Kopp A, Duncan I, Carroll SB (2000). Genetic control and evolution of sexually dimorphic characters in Drosophila. Nature 408: 553–559.
L’Hôte D, Serres C, Laissue P, Oulmouden A, Rogel-Gaillard C, Montagutelli X et al. (2007). Centimorgan-range one-step mapping of fertility traits using interspecific recombinant congenic mice. Genetics 176: 1907–1921.
Leevers SJ, McNeill H (2005). Controlling the size of organs and organisms. Curr Opin Cell Biol 17: 604–609.
Lippman ZB, Semel Y, Zamir D (2007). An integrated view of quantitative trait variation using tomato interspecific introgression lines. Curr Opin Genet Dev 17: 545–552.
Loehlin DW, Oliveira D, Edwards R, Giebel JD, Clark M, Cattani MV et al. (2010). Non-coding changes cause sex-specific wing size differences between closely related species of Nasonia. PLoS Genet (doi:10.1371/journal.pgen.1000821).
Lynch VJ, Wagner GP (2008). Resurrecting the role of transcription factor change in developmental evolution. Evolution 62: 2131–2154.
McCabe J, French V, Partridge L (1997). Joint regulation of cell size and cell number in the wing blade of Drosophila melanogaster. Genet Res 69: 61–68.
McGregor AP, Orgogozo V, Delon I, Zanet J, Srinivasan DG, Payre F et al. (2007). Morphological evolution through multiple cis-regulatory mutations at a single gene. Nature 448: 587–590.
Niehuis O, Judson AK, Gadau J (2008). Cytonuclear genic incompatibilities cause increased mortality in male F-2 hybrids of Nasonia giraulti and N. vitripennis. Genetics 178: 413–426.
Orr HA (2001). The genetics of species differences. Trends Ecol Evol 16: 343–350.
Phillips PC (2008). Epistasis—the essential role of gene interactions in the structure and evolution of genetic systems. Nat Rev Genet 9: 855–867.
Raychoudhury R, Baldo L, Oliveira DCSG, Werren JH (2009). Modes of acquisition of Wolbachia: horizontal transfer, hybrid introgression, and codivergence in the Nasonia species complex. Evolution 63: 165–183.
Raychoudhury R, Desjardins CA, Buellesbach J, Loehlin DW, Grillenberger BK, Beukeboom LW et al. (2010). Behavioural and genetic characteristics of a new species of Nasonia. Heredity (doi:10.1038/hdy.2009.147).
Rütten KB, Pietsch C, Olek K, Neusser M, Beukeboom LW, Gadau J (2004). Chromosomal anchoring of linkage groups and identification of wing size QTL using markers and FISH probes derived from microdissected chromosomes in Nasonia (Pteromalidae: Hymenoptera). Cytogenet Genome Res 105: 126–133.
Stern DL (2000). Perspective: evolutionary developmental biology and the problem of variation. Evolution 54: 1079–1091.
Stern DL, Orgogozo V (2008). The loci of evolution: how predictable is genetic evolution? Evolution 54: 2155–2177.
Weber KE, Greenspan RJ, Chicoine DR, Fiorentino K, Thomas MH, Knight TL (2008). Microarray analysis of replicate populations selected against a wing-shape correlation in Drosophila melanogaster. Genetics 178: 1093–1108.
Werren JH, Perrot-Minnot M-J (1999). An Antennapedia-like mutation in Nasonia vitripennis. J Hered 90: 319–320.
Werren JH, Loehlin D (2009). The parasitoid wasp Nasonia: an emerging model system with haploid male genetics. Cold Spring Harb Protoc (doi: 10.1101/pdb.emo134).
Werren JH, Richards S, Desjardins CA, Niehuis O, Gadau J, Colbourne JK et al. (2010). Functional and evolutionary insights from the genomes of three parasitoid Nasonia species. Science 327: 343.
Weston RF, Qureshi I, Werren JH (1999). Genetics of a wing size difference between two Nasonia species. J Evol Biol 12: 586–595.
Williams TM, Selegue JE, Werner T, Gompel N, Kopp A, Carroll SB (2008). The regulation and evolution of a genetic switch controlling sexually dimorphic traits in Drosophila. Cell 134: 610–623.
Zwaan BJ, Azevedo RBR, James AC, Van ‘T Land J, Partridge L (2000). Cellular basis of wing size variation in Drosophila melanogaster: a comparison of latitudinal clines on two continents. Heredity 84: 338–347.
Acknowledgements
We thank Oliver Niehuis and Juergen Gadau for sharing a pre-release genome map and Victor Anaya and Stephen Richards for sharing their indel datasets. We also thank Michael Clark, Chris Desjardins, and reviewers for helpful suggestions on the manuscript. This work was supported by NIH grant 5R01GM070026-04 to JHW.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Heredity website (http://www.nature.com/hdy)
Supplementary information
Rights and permissions
About this article
Cite this article
Loehlin, D., Enders, L. & Werren, J. Evolution of sex-specific wing shape at the widerwing locus in four species of Nasonia. Heredity 104, 260–269 (2010). https://doi.org/10.1038/hdy.2009.146
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2009.146
Keywords
This article is cited by
-
Quantitative genetics of wing morphology in the parasitoid wasp Nasonia vitripennis: hosts increase sibling similarity
Heredity (2020)
-
Early memory in the parasitoid wasp Nasonia vitripennis
Journal of Comparative Physiology A (2015)
-
The genetic basis of interspecies host preference differences in the model parasitoid Nasonia
Heredity (2010)
-
Behavioral and genetic characteristics of a new species of Nasonia
Heredity (2010)
