
Abstract
The Ca2+ channel-binding domain 3 (CBD3) peptide, derived from the collapsin response mediator protein 2 (CRMP-2), is a recently discovered voltage-gated Ca2+ channel (VGCC) blocker with a preference for CaV2.2. Rodent administration of CBD3 conjugated to cell penetrating motif TAT (TAT-CBD3) has been shown to reduce pain behavior in inflammatory and neuropathic pain models. However, TAT-CBD3 analgesia has limitations, including short half-life, lack of cellular specificity and undesired potential off-site effects. We hypothesized that these issues could be addressed by expressing CBD3 encoded by high-expression vectors in primary sensory neurons. We constructed an adeno-associated viral (AAV) vector expressing recombinant fluorescent CBD3 peptide and injected it into lumbar dorsal root ganglia (DRGs) of rats before spared nerve injury (SNI). We show that selective expression of enhanced green fluorescent protein (EGFP)-CBD3 in lumbar 4 (L4) and L5 DRG neurons and their axonal projections results in effective attenuation of nerve injury-induced neuropathic pain in the SNI model. We conclude that AAV-encoded CBD3 delivered to peripheral sensory neurons through DRG injection may be a valuable approach for exploring the role of presynaptic VGCCs and long-term modulation of neurotransmission, and may also be considered for development as a gene therapy strategy to treat chronic neuropathic pain.
Similar content being viewed by others


Introduction
Peripheral nerve injury often leads to persistent neuropathic pain through altered function of primary sensory neurons and central nervous system pain pathways.1 Voltage-gated Ca2+ channels (VGCCs) have a key role in upregulated sensory neuron excitability and enhanced transmission of pain signals in neuropathic pain.2 Pharmacological and genetic studies implicate N-type Ca2+ channels (CaV2.2) as the principal mediator of pain transmission in the dorsal horn,3, 4 and inhibition of CaV2.2 is an emerging strategy in pain treatment.5 T-type low-voltage-activated Ca2+ channels additionally contribute to neuropathic pain by elevating sensory neuron excitability after nerve injury.6
The axon/dendrite specification protein collapsin response mediator protein 2 (CRMP-2) has recently been shown to interact with CaV2.2, and thereby enhances CaV2.2 cell membrane trafficking and facilitates synaptic transmission.7, 8 CaV2.2 membrane trafficking induced by interaction with CRMP-2 and CaV2.2 can be effectively disrupted by a short peptide corresponding to a 15 amino-acid region of CRMP-2, designated as Ca2+ channel-binding domain 3 (CBD3).8, 9 Importantly, rodent administration by intraperitoneal or systemic injection of CBD3 peptide conjugated to the cell-penetrating motif of the HIV transduction domain protein TAT (TAT-CBD3) has been shown to reduce hypersensitive behavior in chemical-induced inflammatory and neuropathic pain models.9, 10, 11, 12 Functional evaluations show that TAT-CBD3 reduces CaV2.2-mediated currents by inhibiting CRMP-2 binding to CaV2.2, and also reduces T-type currents, which together result in reduced nociceptor excitability, diminished neuropeptide release from sensory neurons and lower excitatory synaptic transmission.11 These observations suggest that CBD3 has the potential for development as a therapeutic agent in pain management. However, the analgesia produced by systematic application of TAT-CBD3 is hampered by its inherent instability, short half-life and other characteristics of systemic peptide therapy, such as lack of anatomic and cellular specificity, as well as the potential for undesired off-target effects triggered by repeated dosing that is necessitated by chronic treatment. In addition, while effective in reducing non-traumatic neuropathic pain behavior, TAT-CBD treatment had no effect on mechanical hypersensitivity in traumatic neuropathic pain.10
We hypothesize that the efficacy and safety could be significantly improved by anatomically restricted expression of therapeutic CBD3 peptide encoded by high-expression vectors in the involved pain pathway. Dorsal root ganglion (DRG) neurons become hyperexcitable after peripheral nerve injury,13, 14 and are the presynaptic neurons at the critical first synapse in the dorsal horn, making them an attractive target for pharmacotherapy. Recombinant adeno-associated viruses (AAVs) have proven to be highly useful vectors for transferring genes into DRG neurons with high efficacy and minimal side effects.15 We have recently optimized a direct microinjection technique to reliably and safely deliver AAV selectively into individual DRGs of rats, leading to efficient, prolonged and segmental transgene expression in DRG neurons and their central processes and presynaptic terminals in the superficial lamina of the spinal cord dorsal horn.16 In a clinical setting, DRG injection is easy to achieve and harmless.17
The present study was designed to test the efficacy of an AAV vector encoding recombinant fluorescent CBD3 injected into lumbar DRGs proximal to peripheral nerve damage. We hypothesized that this would produce a therapeutic level of CBD3 continuously and selectively in the DRG neurons and their central presynaptic terminals, resulting in analgesic neuromodulation. Using the rat spared nerve injury (SNI) model of persistent peripheral neuropathic pain, we have found that selective expression of enhanced green fluorescent protein (EGFP)-CBD3 in lumbar 4 (L4) and L5 DRG neurons and their central and peripheral axonal processes prevents nerve injury-induced neuropathic pain.
Results
Targeted expression of fluorescent CBD3 in the peripheral sensory neurons
Short in vivo half-life has been a general challenge for TAT-conjugated drugable small interfering peptides (designated as peptide aptamers),18, 19, 20 including TAT-CBD3, which is unstable and produces only transient pain relief.10 Scaffold proteins such as GFP have been successfully used to provide a framework for the expression stable peptide aptamers.21, 22 We therefore constructed self-complementary recombinant AAV6 vector expressing CBD3 fused to fluorescent protein EGFP (hereafter referred to as AAV6-EGFP-CBD3). In this construct (Supplementary Figure S1), EGFP provides both a stable scaffold and a fluorescent tag for identifying neurons expressing the CBD3 peptide aptamer (hereafter referred to as EGFP-CBD3). The AAV6 serotype was chosen because we have found that this serotype provides efficient gene transfer to the full range of DRG neurons including the nociceptive sub-population and their axonal terminals.23 Both the L4 and L5 DRGs were injected with either AAV6-EGFP-CBD3 or AAV6-EGFP (5.0 × 109 viral particles each). After 2 weeks, nerve injury was induced by SNI surgery. Four weeks following this, immunohistochemistry characterization (Figures 1a–c) demonstrated successful transduction by AAV6-EGFP-CBD3 of DRG neuronal sub-populations including small peptidergic nociceptive neurons characterized by calcitonin gene-related peptide (CGRP) immunoreactivity (ir), small non-peptidergic neurons characterized by isolectin B4 (IB4) binding and large-sized myelinated neurons characterized by NF-200-ir.

Expression of fluorescent CBD3 in sensory neurons. DRG sections from rats in which AAV6-EGFP-CBD3 was injected 6 weeks previously and SNI traumatic nerve injury was performed 2 weeks thereafter were immunostained with antibodies to EGFP as well as CGRP (a), IB4 (b) or NF-200 (c). Arrowheads point to examples of co-labeled neurons. Spinal cord sections show EGFP-CBD3 expression (d, highlighted area magnified in (e) with enumerated laminae). No colocalization is observed with NeuN staining of dorsal horn neuronal somata (f, magnified in the inset, showing synaptic varicosities of transduced fibers). Sensory neuron fibers in the dorsal horn show cluster with the synaptic marker synaptophysin (g, magnified in the inset). The EGFP-CBD3 signal was also observed in sciatic nerve (h). Scale bars: 100 μm for all images except 50 μm for inset images. Western analysis (i) of HEK293T cell lysates following plasmid transfection show EGFP immunoreactivity at distinct molecular weights (MWs) for expressed EGFP (lane 2) versus EGFP-CBD3 (lane 3) as positive controls. DRG homogenates show no immunoreactivity in DRGs contralateral from the injection (lane 4), and appropriate MW in homogenates from DRGs injected with AAV6-EGFP-CBD3 (lanes 5 and 6) or AAV6-EGFP (lanes 7 and 8). Lane 1 shows marker protein standards (M; MagicMark, Life Technologies). Arrows point to the expected size bands for EGFP-CBD3 and EGFP (top panel), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH, bottom panel) as a loading control.
The in vivo transduction rate for AAV6-EGFP-CBD3 was 31±3.5% of total neuronal profiles (positive for β3-tubulin) within sections showing the entire ganglion (n=3 DRGs, 5 sections per DRG). Examination of the corresponding spinal cord revealed abundant EGFP-CBD3-ir fibers in the dorsal horn only ipsilateral to the injections, including in the superficial laminae (Figures 1d and e), evident by colocalization of EGFP with superficial laminae markers, CGRP, IB4 and CaMKII (data not shown), as previously reported.23 EGFP-CBD3-ir was not observed in dorsal horn neuronal somata labeled with a pan neuron-specific marker NeuN (Figure 1f), but was clustered with the presynaptic marker synaptophysin (Figure 1g). These findings indicate that the transgene product (EGFP-CBD3) extends to the central presynaptic terminals but is not transported across central synapses. EGFP-CBD3-ir was also observed in sciatic nerve (Figure 1h), indicative of transport of EGFP-CBD3 to the peripheral processes of transduced sensory neurons. Similar patterns of EGFP-ir in the DRG neurons and their axonal projections were detected following AAV6-EGFP injection (data not shown), as previously reported.23 Western blots of DRG tissue homogenates identified the EGFP-CBD3 fusion protein based on the size disparity between EGFP-CBD3 and EGFP, indicating stability of EGFP-CBD3 structure during intraganglionic expression (Figure 1i). Together, these observations indicate that intraganglionic microinjection of AAV6 encoding CBD3 produces long-term expression of fluorescent CBD3 selectively in DRG neurons and their central and peripheral axonal terminals.
Effect of EGFP-CBD3 on Ca2+ currents
Prior observations have demonstrated a reduction in N-type ICa after transient exposure of sensory neurons to CBD3.9 We therefore examined the efficacy of sustained expression in sensory neurons of the EGFP-CBD3 transgene on current sensitive to the selective N-type ICa blocker ω-Conotoxin-GVIA (GVIA, 200 nM).24 Using a whole-cell patch-clamp approach and conditions to isolate ICa, transduced neurons identified by EGFP fluorescence (Figure 2a) were held at −60 mV, hyperpolarized to −100 mV for 1 s, and then depolarized for 200 ms to +40 mV in 10 mV increments. Peak ICa, which occurred at −10 to 0 mV, did not differ between neurons expressing EGFP (158±11 pA/pF, n=10) and EGFP-CBD3 (134±18 pA/pF, n=9; P=0.26). During subsequent application of GVIA, neurons were depolarized by test pulses to 0 mV, which fully activates N-type ICa in sensory neurons24 (Figures 2b and c). Trace subtraction demonstrated that current sensitive to GVIA was reduced in neurons expressing EGFP-CBD3 compared with EGFP alone (P<0.05; Figure 2d), and that this N-type current represented a reduced fraction of the total ICa in neurons expressing EGFP-CBD3 (26±4%) compared with EGFP alone (42±3%, P<0.01), which indicate the efficacy of the EGFP-CBD3 construct in reducing N-type ICa.

Electrophysiological properties of neurons expressing EGFP-CBD3. Transduced neurons were identified after dissociation by EGFP epifluorescence (a, bottom image; top image: phase-contrast). N-type current was identified as the difference between current at baseline (BL) and after application of ω-Conotoxin-GVIA (GVIA, 200 nM) in response to depolarizations from −100 mV to VTest of 0 mV (b, sample traces; c, time course for group average) for neurons expressing EGFP (n=10) or EGFP-CBD3 (n=9). Summary data (d) show a significant depression of N-type current in neurons expressing EGFP-CBD3 compared with EGFP (*P<0.05). T-type current traces triggered by depolarizations from VH −100 mV to VTest of −30 mV (e) after incubation (20 min) with R-type current blocker SNX-482 (200 nM) and P/Q-type current blocker ω-Conotoxin MVIIC (200 nM), and addition of L-type current blocker nimodipine (5 μM) and GVIA (200 nM) to the recording bath. Summary data (f) show a significant depression of T-type current in neurons expressing EGFP-CBD3 compared with EGFP (**P<0.01).
CBD3 has also been noted to suppress low-voltage-activated T-type current,11 which increases after nerve injury and contributes to excitability.25 We therefore determined the amount of T-type current in transduced neurons using an established method in which this current is selectively triggered by depolarizations from a holding potential of −100 mV to a test potential of −30 mV during blockade of high-voltage-activated currents (Figure 2e), and correcting the current peak by subtracting the sustained component.25 Neurons expressing EGFP-CBD3 developed less T-type current than neurons expressing EGFP alone (P<0.01; Figure 2f). Non-transduced neurons had T-type current (29.8±4.0 pA/pF, n=10) that did not differ from currents in neurons expressing EGFP alone. Thus, chronic expression of the EGFP-CBD3 construct has efficacy in blocking T-type current as well as N-type current, replicating findings from studies that examined acute application of TAT-CBD3.11
EGFP-CBD3 expression in DRG neurons attenuates SNI-induced neuropathic pain
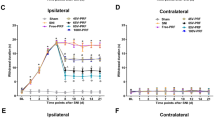
Since both N-type and T-type ICa have been shown to participate in pain through regulation of sensory neuron excitability and neurotransmission in pain pathways, we next evaluated whether sustained neuronal expression of EGFP-CBD3 altered pain processing under baseline conditions and following nerve injury. Our previous studies show that transgene expression begins within 1 week after intraganglionic delivery of AAV6, and leads to robust transgene expression at 2 weeks.23 Therefore, nerve injury was performed by SNI 2 weeks following injection of either AAV6-EGFP-CBD3 or AAV6-EGFP to both L4 and L5 DRGs. Sensitivity to cutaneous mechanical and cold stimulation was evaluated before and after DRG injection of the vectors and continued for 6 weeks after the nerve injury. These specific sensory modalities were tested since increased sensitivity to mechanical and cold stimuli has been reported as the most representative of the injury phenotype following SNI.26 During the 2 weeks between vector injection and SNI, no significant changes were observed in responsiveness to mechanical or cold stimuli (Figures 3a–c). In other rats without nerve injury surgery, such normal sensory behavior continued throughout the full testing period of 4 weeks after injection (n=6 for each vector; data not shown). This indicates that EGFP-CBD3 expression had no significant effect on the normal sensory function for these modalities. Following SNI surgery, all animals injected with AAV6-EGFP developed a significant mechanical allodynia to normally innocuous punctate mechanical stimulation (von Frey) and hyperalgesia to noxious mechanical stimulation (pin), as well as elevated sensitivity to cold. During the initiation and maintenance stages of chronic pain following SNI, these behavioral changes were significantly attenuated in animals injected with AAV6-EGFP-CBD3 compared with the animals injected with AAV6-EGFP control vector (Figures 3d–f). Thus, EGFP-CBD3 reduced SNI-induced mechanical hyperalgesia (pin test) by 64%, whereas SNI-induced sensitization to von Frey testing was reduced by 28%, and cold sensitization was reduced by 52%. These results indicate that sustained expression of the EGFP-CBD3 selectively in the primary sensory neurons of the L4 and L5 DRGs significantly attenuates injury-induced persistent neuropathic pain.

Therapeutic efficacy and safety of intraganglionic AAV-encoded CBD3 in SNI-induced neuropathic pain. Left panels show the time courses for the group averages of sensitivity to innocuous punctate mechanical stimulation (von Frey, a), hyperalgesia behavior after touch with a pin (Pin, b), and sensitivity to acetone stimulation (Cold, c) before and after DRG injection of either AAV6-EGFP (filled circle, b=7) or AAV6-EGFP-CBD3 (open square, n=7). Injection of AAV vector into the fourth and fifth lumbar DRGs was performed immediately after the baseline (BL) behavioral determinations, and spared nerve injury (SNI) was performed immediately after the week 2 determinations. Right panels show averaged area under the curve calculated for each individual for the time period following SNI for von Frey (d), pin (e) and cold (f). Results are means±s.e.m. *P<0.05, **P<0.01, ***P<0.001 for comparisons to BL (a–c) and for comparisons between groups (d–f).
EGFP-CBD3 expression does not reverse SNI-induced inflammatory response
Apart from direct modulation of membrane channel function, it is possible that analgesia induced by EGFP-CBD3 expression may result from reduction in the inflammatory processes in the DRG and spinal cord that follow peripheral nerve injury.27 Conversely, it is also possible that vector injection and/or transgene expression could enhance inflammation, and thereby account for the incomplete efficacy of CBD3 analgesia after SNI. We therefore examined DRGs and spinal cords from injected animals (n=3 for each vector) for Iba1-ir, which identifies microglial activation and macrophage accumulation, and for GFAP-ir, which identifies satellite glial cell and astrocyte activation. Tissue was collected 6 weeks after DRG injection and 4 weeks after SNI. Compared with uninjured animals, the DRGs of SNI rats receiving vehicle, AAV6-EGFP or AAV6-EGFP-CBD3 injections all developed comparable increases in both GFAP-ir and Iba1-ir, as glial rings surrounding neuronal somata (Figure 4a). Similarly, the ipsilateral spinal cords of all SNI animals developed elevated astrocyte and microglial staining with hypertrophic cell bodies, regardless of DRG injections (Figure 4b). These results indicate that the EGFP-CBD3 expression does not suppress nerve injury-triggered inflammatory reactions in the DRG or the spinal cord, nor does it likely contribute to these changes.

EGFP-CBD3 expression does not suppress nerve injury-triggered inflammatory reactions. Representative sections from DRG (a) and spinal cord dorsal horn (b) showing Iba1 (top row) and GFAP (bottom row) immunoreactivity. Tissues are from rats 6 weeks after vehicle injection without injury (first column), 6 weeks after vehicle injection and 4 weeks after SNI (second column), 6 weeks after injection of AAV6-EGFP followed by SNI (third column) and 6 weeks after AAV6-EGFP-CBD3 injection followed by SNI (fourth column). Scale bars: 50 μm, and apply to all images in the panel.
Discussion
Small peptide aptamers targeting protein–protein interactions in pain signaling pathways have recently gained increasing interest as research tools and as potential pharmacological agents for treating chronic pain. Specific peptide motifs derived from endogenous proteins can bind to the cognate sites of target proteins with both high affinity and specificity, resulting in selective interference of the function of their target signaling protein. Unlike strategies such as RNA interference or gene knockout that reduce the production of a target protein, peptide aptamer interference can selectively block particular interactions without eliminating either partner protein per se, resulting in specific ‘functional knockdown’ of a selected signaling pathway. A number of synthetic peptide inhibitors, including CBD3 conjugated to the TAT motif, have shown efficacy for pain caused by various pathological conditions,9, 28, 29 and systemic administration of TAT-mediated analgesic peptides has been proposed as a promising alternative approach for the treatment of chronic pain.20, 30 However, this strategy also has some limitations, including the inability to target specific organs or cells. Another major obstacle in using TAT as a delivery vector is its rapid clearance when administrated in vivo, resulting in total clearance in as rapidly as 2 h after a bolus tail vein injection.31 This rapid metabolism and clearance may account for the transience of the behavioral effects of TAT-mediated analgesic peptide therapy. The high susceptibility of TAT-linked therapeutic peptides to intracellular degradation may additionally limit their potential utility.32 These points argue for an alternate mode of administration.
Direct DRG injection of AAV vectors is a feasible, safe and efficient approach for genetically modifying segmentally restricted populations of peripheral sensory neurons,16, 33 including their central presynaptic terminals.23 We show that a single AAV injection leads to persistent CBD3 expression selectively in DRG neurons and their central and peripheral processes, and provides sustained pain relief with minimal toxicity. These results provide clear evidence of the therapeutic potential of Ca2+ channel modulation by AAV-targeted local biosynthesis of CBD3 in DRGs for treating neuropathic pain.
We chose VGCCs as an initial molecular target due to their central role in regulating neuronal excitability and synaptic transmission. CaV2.2 is the critical source of Ca2+ influx that triggers neurotransmitter release in pain transmission in the spinal cord dorsal horn.34 Furthermore, presynaptic expression of CaV2.2 may increase in chronic pain states35 with resulting increase in dependence of nociceptive pain transmission upon CaV2.2.36 These features make it an ideal target for presynaptic modulation of chronic pain.37 Indeed, the selective CaV2.2 peptide blocker ω-conotoxin MVIIA (ziconotide, Prialt, Elan Pharmaceuticals, San Francisco, CA, USA) is marketed for clinical use by intrathecal infusion, during which intense analgesia can be achieved but at the cost of a very high frequency of brain toxicity-related complications.38 Clearly, limiting CaV2.2 blockade to the presynaptic sensory neurons is a more desirable strategy.
This study was not designed to determine the site of action at which expression of CBD3 within the sensory neuron produces analgesia. However, since both CRMP-2 and CaV2.2 are expressed throughout the sensory neuron, CBD3 disruption of their interaction can occur at multiple sites.7 Diminished ICa at the sensory neuron terminals in the dorsal horn may have a substantial analgesic effect due to the steep dependence of neurotransmitter release upon Ca2+ influx.39 Others have found that CaV2.2 blockers applied to somata of axotomized sensory neurons decrease their spontaneous activity,40 whereas CaV2.2 blockade at the nerve injury site reverses thermal and mechanical hypersensitivity.41 We have also provided evidence that our construct reduces T-type current, which may additionally have a role in quelling pain by reducing the contribution of this current to both neurotransmission39 and peripheral neuronal excitability.42
Although our findings demonstrate sustained relief of traumatic neuropathic pain from AAV-encoded expression of CBD3 in primary sensory neurons, the efficacy is incomplete, for which several explanations are possible. The approximately 30% transduction efficiency leaves a persisting population of sensory neurons with their natural post-injury functional phenotype intact, which may in turn be sufficient to maintain sensitized behavior. Possible improvement in transduction efficiency might be achieved by increasing the viral titer through larger scale production techniques, or by employing new AAV serotypes that are continually becoming available.11 Another possible strategy would be refining the vector design, such as targeting the CBD3 preferentially to the presynaptic membrane.43 An additional hypothesis to explain incomplete efficacy of our construct is insufficient blocking effect of the CBD3 peptide against CaV2.2. Approaches to improve its efficacy could include employing a recently described variant CBD3 sequence with enhanced inherent efficacy,11 or designing the construct to express a concatemerized peptide composed of multiple linked copies of the active sequence.44, 45 It is also noteworthy that CBD3 expression showed no anti-inflammatory efficacy. This is consistent with an interpretation that the anti-hyperalgesic efficacy of CBD3 expression in sensory neurons does not arise for altering primary pathogenic processes such as inflammation that generate the hyperalgesic state. Rather, CBD3 probably influences downstream processes such as neuronal excitation and neurotransmission.
Overall, our findings show that AAV delivery of a small interfering peptide is an efficient strategy for sustained treatment of neuropathic pain. Although pain blockade is incomplete, absolute elimination of chronic pain in a clinical setting is not a requirement for successful treatment. The features of highly accurate anatomic targeting and molecular specificity of the expressed peptide indicate that this approach has the potential for translation into an effective therapeutic approach.
Materials and methods
Animals
Adult male Sprague Dawley rats weighting 125–150 g body weight (Charles River Laboratories, Wilmington, MA, USA) were housed individually in a room maintained at constant temperature (22±0.5 °C) and relative humidity (60±15%) with an alternating 12 h light-dark cycle with free access to water and food throughout the experiments. All procedures were performed with the approval of the Zablocki VA Medical Center Animal Studies Subcommittee and the Medical College of Wisconsin Institutional Animal Care and Use Committee, in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
AAV vectors
The plasmids used for recombinant AAV production were (1) self-complimentary AAV expression plasmid (pscAAV-EGFP) containing a CMV-driven EGFP expression cassette with the simian virus 40 poly(A) signal, flanked by sc AAV2 inverted terminal repeats;23 (2) pRC6 containing AAV2 replication gene and capsid gene from AAV6 (Viromics, Fremont, CA, USA); and (3) pHelper encoding the adenoviral helper genes (Viromics). Construction of the plasmid expressing the EGFP-CBD3 peptide aptamer was achieved by inserting the oligonucleotides encoding CBD3 peptide (ARSRLAELRGVPRGL) into the 3′ of EGFP of pscAAV-EGFP, separated by oligonucleotides encoding a 22 amino-acid linker (SGLRSRAQASNSAVDGTAGPGS) derived from the polylinker regions of pEGFPc1. The resultant plasmid was named as pscAAV-EGFP-CBD3. The recombinant AAV was packaged by triple-plasmid CaPO4 co-transfection of 293T cells. Virions were Optiprep purified, concentrated with Centricon Plus-20 (Regenerated Cellulose 100 000 MWCO; Millipore, Billerica, MA, USA), and titer determined by a PicoGreen method, as reported previously.23 The resulting vectors, AAV6-EGFP-CBD3 and AAV6-EGFP, possessed titers of 5.5 × 1012 and 2.1 × 1013 genome copies per ml (GC ml−1), respectively. Infectivity was estimated by evaluating the percentage of EGFP-positive HEK293T cells 48 h after transduction of either AAV6-EGFP-CBD3 or AAV6-EGFP at a multiplicity of infection of 10 000. By immunoblotting, the EGFP-CBD3 chimeric protein was detected in lysates of HEK293T cells infected with AAV6-EGFP-CBD3 with a mass that is ∼10 kDa larger than EGFP (Supplementary Figure S1a). Comparable purity of AAV6-EGFP-CBD3 and AAV6-EGFP was confirmed by silver staining showing dominant viral Vp1, Vp2 and Vp3 proteins (95% for AAV6-EGFP and 88% for AAV6-EGFP-CBD3) in gel (Supplementary Figure S1b).
Microinjection of AAV vectors into DRGs
AAV vector was microinjected into right L4 and L5 DRGs using previously described techniques.16 Briefly, the surgically exposed intervertebral foramen was minimally enlarged by removal of laminar bone. Injection was performed through a micropipette that was advanced ∼100 μm into the ganglion. Rats received L4 and L5 DRG injections of either AAV6-EGFP-CBD3 or AAV6-EGFP (one vector per rat), consisting of 2 μl with adjusted titers containing a total of 5.0 × 109 genome containing viral particles. Injection was performed over a 5-min period using a Nanoliter 2000 microprocessor-controlled injector (World Precision Instruments, Sarasota, FL, USA). Removal of the pipette was delayed for an additional 5 min to minimize the extrusion of the injectate. Following the injection and closure of overlying muscle and skin, the animals were returned to the animal house where they remained for 4 weeks.
Spared nerve injury
SNI produces consistent, reliable and long-lasting post-traumatic neuropathic pain behaviors featuring mechanical allodynia and hyperalgesia, and hypersensitivity to cold induced by acetone application.26, 46 For SNI surgery, animals were anesthetized by exposure to isoflurane (1–3%). The surgical field (right leg) was shaved and disinfected. An incision (∼2 cm) was made on the lateral mid-thigh and the underlying muscles separated to expose the sciatic nerve, and the tibial and common peroneal were individually ligated with 6.0 sutures and cut distally to the ligature, and 2–3 mm of each nerve was removed distal of the ligation. The sural nerve was preserved and contact with it was avoided. Muscle and skin were closed using 4.0 monofilament nylon sutures.
Pain behavioral evaluation
Behavioral tests were carried out as previously described.16 Mechanical withdrawal threshold testing (von Frey) was performed using calibrated monofilaments (Patterson Medical, Bolingbrook, IL, USA). Briefly, beginning with the 2.8-g filament, filaments were applied with just enough force to bend the fiber and held for 1 s. If a response was observed, then the next smaller filament was applied, and if no response was observed, then the next larger was applied, until a reversal occurred, defined as a withdrawal after a previous lack of withdrawal, or vice versa. Following a reversal event, four more stimulations were performed following the same pattern. The forces of the filaments before and after the reversal, and the four filaments applied following the reversal, were used to calculate the 50% withdrawal threshold. Rats not responding to any filament were assigned a score of 25 g. Noxious punctate mechanical stimulation (pin test) was performed using the point of a 22-g spinal anesthesia needle that was applied to the center of the hindpaw with enough force to indent the skin but not puncture it. Five applications were separated by at least 10 s, which was repeated after 2 min, making a total of 10 touches. For each application, the induced behavior was either a very brisk, simple withdrawal with immediate return of the foot to the cage floor, or a sustained elevation with grooming that included licking and chewing, and possibly shaking, which lasted at least 1 s.47 This hyperalgesic behavior is specifically associated with place avoidance.48 Hyperalgesia was quantified by tabulating hyperalgesia responses as a percentage of total touches.
Immunohistochemistry and imaging
Staining of paraffin-embedded sections was performed by a standard fluorescent protocol, as previously described.23 Sections were immunolabeled with the primary antibodies against GFP (monoclonal GFP, 1:400, Santa Cruz Biotechnology, SCB, Santa Cruz, CA, USA; rabbit polyclonal GFP, 1:400, Cell Signaling, Danvers, MA, USA), monoclonal β3-tubulin (1:500, SCB), monoclonal CGRP (1:600, SCB), rabbit polyclonal synaptophysin (1:500, SCB), rabbit polyclonal CaMKII (SCB, 1:600), monoclonal NF200 (1:1000, Sigma-Aldrich, St Louis, MO, USA), monoclonal NeuN (1:200, Millipore), rabbit polyclonal GFAP (1:4000, Dako, Carpinteria, CA, USA), rabbit polyclonal Iba1(Dako, 1:2000), and biotinylated-isolectin-B4 (IB4, 10 μg ml−1, Life Technologies, Carlsbad, CA, USA) binding, with BSA replacement of first antibody as the negative control. The appropriate fluorophore-conjugated (Alexa 488 or Alexa 594) secondary antibodies (Jackson Immunoresearch, West Grove, PA, USA) were used to reveal the primary antibodies. The sections were examined and images captured using a Nikon TE2000-S fluorescence microscope (NIKON Instruments, Melville, NY, USA) with filters suitable for selectively detecting the green and red fluorescence, and an Optronics QuantiFire digital camera.
Western blots
Briefly, homogenates from DRGs injected with AAV6-EGFP-CBD3 or AAV6-EGFP were loaded onto SDS–PAGE, transferred, and probed with a polyclonal rabbit anti-GFP antibody (1:1000; Cell Signaling). Immunoreactive proteins were detected by enhanced chemiluminescence (Pierce, Rockford, IL, USA) after incubation with HRP-conjugated anti-rabbit IgG (1:2000, SCB). Lysate of HEK293T cells transduced with AAV6-EGFP-CBD3 or AAV6-EGFP was extracted as the positive controls. GAPDH was used as a loading control.
Whole-cell patch-clamp recording
To determine the effect of EGFP-CBD3, recordings were made from neurons dissociated from the L4 and L5 DRGs of uninjured rats that had been injected with either AAV6-EGFP or AAV6-EGFP-CBD3. Following decapitation under deep isoflurane anesthesia, ganglia were removed and placed in a 35-mm dish containing Ca2+/Mg2+-free, cold HBBS (Life Technologies) and cut into 4–6 pieces that were incubated in 0.5 mg ml−1 liberase TM (Roche, Indianapolis, IN, USA) in DMEM/F12 with glutaMAX (Life Technologies) for 30 min at 37 °C, followed by 1 mg ml−1 trypsin (Sigma-Aldrich) and 150 Kunitz units per ml DNase (Sigma-Aldrich) for another 10 min. After addition of trypsin inhibitor (Type II, Sigma-Aldrich), tissues were centrifuged, lightly triturated in neural basal media (1 × ) (Life Technologies) containing 2% (v:v) B27 supplement (50 × ) (Life Technologies), 0.5 mM glutamine (Sigma-Aldrich), 0.05 mg ml−1 gentamicin (Life Technologies) and 10 ng ml−1 nerve growth factor 7S (Alomone Labs Ltd., Jerusalem, Israel). Cells were then plated onto poly-L-lysine (70–150 kDa, Sigma-Aldrich) coated coverslips and cultured at 37 °C in 5% CO2. All cells were studied 3–8 h after dissociation. Medium-sized neurons (27±0.4 μm) were used for this study.
Electrodes with a resistance of 2–4 MΩ were pulled from borosilicate glass (Garner Glass Co., Claremont, CA, USA) using a micropipette puller (P-97 Sutter Instrument Co, Novato, CA, USA) and fire polished. Recording was performed in the whole-cell configuration with an Axopatch 700B amplifier (Molecular Devices, Sunnyvale, CA, USA). After whole-cell configuration was established, electrical compensation for the cell membrane capacitance and series resistance were initiated. Access resistance was typically between 5 and 7 MΩ and was 80–90% compensated. Since peak currents measured in our experiment were around 3 nA, voltage errors introduced by the residual uncompensated access resistance (<1.0 MΩ) were <3 mV and could not introduce major errors. Neurons with >10 MΩ access resistance after breakthrough were discarded. Experiments were performed 5 min after breakthrough, and at room temperature (∼25 °C). Signals were filtered at 2 kHz through a 4-pole Bessel filter, and digitized at 10 kHz with a Digidata 1440 A A/D interface (Molecular Devices). Seals were achieved in modified Tyrode’s consisting of (in mM) NaCl 140, KCl 4, CaCl2 2, MgCl2 2, D-glucose 10, and 4-(2-hydroxyethyl)-1-piperazine-1-ethanesulfonic acid (HEPES) 10, at a pH of 7.4, and with an osmolarity of 300 mOsm. Currents through VGCCs were recorded using an extracellular solution consisting of BaCl2 2, 4-aminophyridine 1, HEPES 10, tetraethylammonium chloride (TEA-Cl) 140, and HEPES 10, at a pH of 7.4, and with an osmolarity of 300 mOsm. The internal pipette solution contained CsCl 110, TEACl 20, Mg-ATP 4, Na-GTP 0.3, ethylene glycol-bis(2-amino-ethylether)-N,N, N’, N’-tetra-acetic acid (EGTA) 11, CaCl2 1, MgCl2 1, 4 Mg-ATP, 0.4 Li4-GTP and HEPES 10, at a pH of 7.2, and with an osmolarity of 300 mOsm. To selectively record low-voltage-activated T-type currents, we used a pipette solution that contained fluoride to facilitate high-voltage-activated ICa rundown.49 This solution contained tetramethylammonium hydroxide (TMA-OH) 135, EGTA 10, HEPES 40 and MgCl2 2, adjusted to pH 7.2 with hydrofluoric acid, and with an osmolarity of 300 mOsm. Data from whole-cell ICa recordings were evaluated with Axograph X 1.3.5 (AxoGraph Scientific, Sydney, Australia), with which peak inward currents and charge transfer were measured. To correct for cell size, inward currents are expressed as a function of cell capacitance (pA/pF).
Statistical analyses
Results are reported as mean and variability as s.e.m. A probability of P<0.05 was considered as significant. For continuous variables (von Frey), within-group testing was performed using one-way ANOVA with repeated measures, with separate tests for each vector, both before and after injury. Comparisons to baseline for each group were performed using Dunnet’s test. For non-continuous variables (pin and cold), the non-parametric Friedman’s ANOVA was used, with paired comparisons to baseline with Dunn’s test. For comparisons between groups, data before and after injury for each vector were transformed to area under the curve. Area under the curve values were compared between vectors both before and after injury by the Mann–Whitney U test. All analyses were performed using Prism 6 (GraphPad Software, La Jolla, CA, USA).
References
Costigan M, Scholz J, Woolf CJ . Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci 2009; 32: 1–32.
Park J, Luo ZD . Calcium channel functions in pain processing. Channels (Austin) 2010; 4: 510–517.
Saegusa H, Kurihara T, Zong S, Kazuno A, Matsuda Y, Nonaka T et al. Suppression of inflammatory and neuropathic pain symptoms in mice lacking the N-type Ca2+ channel. EMBO J 2001; 20: 2349–2356.
Saegusa H, Matsuda Y, Tanabe T . Effects of ablation of N- and R-type Ca(2+) channels on pain transmission. Neurosci Res 2002; 43: 1–7.
Pexton T, Moeller-Bertram T, Schilling JM, Wallace MS . Targeting voltage-gated calcium channels for the treatment of neuropathic pain: a review of drug development. Expert Opin Investig Drugs 2011; 20: 1277–1284.
Todorovic SM, Jevtovic-Todorovic V . Neuropathic pain: role for presynaptic T-type channels in nociceptive signaling. Pflugers Arch 2013; 465: 921–927.
Chi XX, Schmutzler BS, Brittain JM, Wang Y, Hingtgen CM, Nicol GD et al. Regulation of N-type voltage-gated calcium channels (Cav2.2) and transmitter release by collapsin response mediator protein-2 (CRMP-2) in sensory neurons. J Cell Sci 2009; 122: 4351–4362.
Brittain JM, Piekarz AD, Wang Y, Kondo T, Cummins TR, Khanna R . An atypical role for collapsin response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. J Biol Chem 2009; 284: 31375–31390.
Brittain JM, Duarte DB, Wilson SM, Zhu W, Ballard C, Johnson PL et al. Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca(2) channel complex. Nat Med 2011; 17: 822–829.
Wilson SM, Brittain JM, Piekarz AD, Ballard CJ, Ripsch MS, Cummins TR et al. Further insights into the antinociceptive potential of a peptide disrupting the N-type calcium channel-CRMP-2 signaling complex. Channels (Austin) 2011; 5: 449–456.
Piekarz AD, Due MR, Khanna M, Wang B, Ripsch MS, Wang R et al. CRMP-2 peptide mediated decrease of high and low voltage-activated calcium channels, attenuation of nociceptor excitability, and anti-nociception in a model of AIDS therapy-induced painful peripheral neuropathy. Mol Pain 2012; 8: 54.
Ripsch MS, Ballard CJ, Khanna M, Hurley JH, White FA, Khanna R . A peptide uncoupling crmp-2 from the presynaptic ca(2+) channel complex demonstrates efficacy in animal models of migraine and aids therapy-induced neuropathy. Transl Neurosci 2012; 3: 1–8.
Sapunar D, Ljubkovic M, Lirk P, McCallum JB, Hogan QH . Distinct membrane effects of spinal nerve ligation on injured and adjacent dorsal root ganglion neurons in rats. Anesthesiology 2005; 103: 360–376.
Chung JM, Chung K . Importance of hyperexcitability of DRG neurons in neuropathic pain. Pain Pract 2002; 2: 87–97.
Beutler AS . AAV provides an alternative for gene therapy of the peripheral sensory nervous system. Mol Ther 2010; 18: 670–673.
Fischer G, Kostic S, Nakai H, Park F, Sapunar D, Yu H et al. Direct injection into the dorsal root ganglion: technical, behavioral, and histological observations. J Neurosci Methods 2011; 199: 43–55.
Pfirrmann CW, Oberholzer PA, Zanetti M, Boos N, Trudell DJ, Resnick D et al. Selective nerve root blocks for the treatment of sciatica: evaluation of injection site and effectiveness--a study with patients and cadavers. Radiology 2001; 221: 704–711.
Schwartz JJ, Zhang S . Peptide-mediated cellular delivery. Curr Opin Mol Ther 2000; 2: 162–167.
Brooks H, Lebleu B, Vives E . Tat peptide-mediated cellular delivery: back to basics. Adv Drug Deliv Rev 2005; 57: 559–577.
Tao F, Johns RA . Tat-mediated peptide intervention in analgesia and anesthesia. Drug Dev Res 2010; 71: 99–105.
Abedi MR, Caponigro G, Kamb A . Green fluorescent protein as a scaffold for intracellular presentation of peptides. Nucleic Acids Res 1998; 26: 623–630.
Hoppe-Seyler F, Crnkovic-Mertens I, Tomai E, Butz K . Peptide aptamers: specific inhibitors of protein function. Curr Mol Med 2004; 4: 529–538.
Yu H, Fischer G, Ferhatovic L, Fan F, Light AR, Weihrauch D et al. Intraganglionic AAV6 results in efficient and long-term gene transfer to peripheral sensory nervous system in adult rats. PLoS One 2013; 8: e61266.
McCallum JB, Wu HE, Tang Q, Kwok WM, Hogan QH . Subtype-specific reduction of voltage-gated calcium current in medium-sized dorsal root ganglion neurons after painful peripheral nerve injury. Neuroscience 2011; 179: 244–255.
Jagodic MM, Pathirathna S, Joksovic PM, Lee W, Nelson MT, Naik AK et al. Upregulation of the T-type calcium current in small rat sensory neurons after chronic constrictive injury of the sciatic nerve. J Neurophysiol 2008; 99: 3151–3156.
Decosterd I, Woolf CJ . Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 2000; 87: 149–158.
Watkins LR, Maier SF . Beyond neurons: evidence that immune and glial cells contribute to pathological pain states. Physiol Rev 2002; 82: 981–1011.
Liu XJ, Gingrich JR, Vargas-Caballero M, Dong YN, Sengar A, Beggs S et al. Treatment of inflammatory and neuropathic pain by uncoupling Src from the NMDA receptor complex. Nat Med 2008; 14: 1325–1332.
Wilson SM, Schmutzler BS, Brittain JM, Dustrude ET, Ripsch MS, Pellman JJ et al. Inhibition of transmitter release and attenuation of anti-retroviral-associated and tibial nerve injury-related painful peripheral neuropathy by novel synthetic Ca2+ channel peptides. J Biol Chem 2012; 287: 35065–35077.
Borghouts C, Kunz C, Groner B . Peptide aptamer libraries. Comb Chem High Throughput Screen 2008; 11: 135–145.
Polyakov V, Sharma V, Dahlheimer JL, Pica CM, Luker GD, Piwnica-Worms D . Novel Tat-peptide chelates for direct transduction of technetium-99 m and rhenium into human cells for imaging and radiotherapy. Bioconjug Chem 2000; 11: 762–771.
Cunha FM, Berti DA, Ferreira ZS, Klitzke CF, Markus RP, Ferro ES . Intracellular peptides as natural regulators of cell signaling. J Biol Chem 2008; 283: 24448–24459.
Mason MR, Ehlert EM, Eggers R, Pool CW, Hermening S, Huseinovic A et al. Comparison of AAV serotypes for gene delivery to dorsal root ganglion neurons. Mol Ther 2010; 18: 715–724.
Rycroft BK, Vikman KS, Christie MJ . Inflammation reduces the contribution of N-type calcium channels to primary afferent synaptic transmission onto NK1 receptor-positive lamina I neurons in the rat dorsal horn. J Physiol 2007; 580: 883–894.
Cizkova D, Marsala J, Lukacova N, Marsala M, Jergova S, Orendacova J et al. Localization of N-type Ca2+ channels in the rat spinal cord following chronic constrictive nerve injury. Exp Brain Res 2002; 147: 456–463.
Matthews EA, Dickenson AH . Effects of spinally delivered N- and P-type voltage-dependent calcium channel antagonists on dorsal horn neuronal responses in a rat model of neuropathy. Pain 2001; 92: 235–246.
Yaksh TL . Calcium channels as therapeutic targets in neuropathic pain. J Pain 2006; 7: S13–S30.
Schmidtko A, Lotsch J, Freynhagen R, Geisslinger G . Ziconotide for treatment of severe chronic pain. Lancet 2010; 375: 1569–1577.
Jacus MO, Uebele VN, Renger JJ, Todorovic SM . Presynaptic Cav3.2 channels regulate excitatory neurotransmission in nociceptive dorsal horn neurons. J Neurosci 2012; 32: 9374–9382.
Liu X, Zhou JL, Chung K, Chung JM . Ion channels associated with the ectopic discharges generated after segmental spinal nerve injury in the rat. Brain Res 2001; 900: 119–127.
Xiao WH, Bennett GJ . Synthetic omega-conopeptides applied to the site of nerve injury suppress neuropathic pains in rats. J Pharmacol Exp Ther 1995; 274: 666–672.
Nelson MT, Joksovic PM, Perez-Reyes E, Todorovic SM . The endogenous redox agent L-cysteine induces T-type Ca2+ channel-dependent sensitization of a novel subpopulation of rat peripheral nociceptors. J Neurosci 2005; 25: 8766–8775.
Dubiel SM, Cieslak J, Sturhahn W, Sternik M, Piekarz P, Stankov S et al. Vibrational properties of alpha- and sigma-phase Fe-Cr alloy. Phys Rev Lett 2010; 104: 155503.
Ji Y, Li B, Reed TD, Lorenz JN, Kaetzel MA, Dedman JR . Targeted inhibition of Ca2+/calmodulin-dependent protein kinase II in cardiac longitudinal sarcoplasmic reticulum results in decreased phospholamban phosphorylation at threonine 17. J Biol Chem 2003; 278: 25063–25071.
Sato AK, Viswanathan M, Kent RB, Wood CR . Therapeutic peptides: technological advances driving peptides into development. Curr Opin Biotechnol 2006; 17: 638–642.
Pertin M, Gosselin RD, Decosterd I . The spared nerve injury model of neuropathic pain. Methods Mol Biol 2012; 851: 205–212.
Hogan Q, Sapunar D, Modric-Jednacak K, McCallum JB . Detection of neuropathic pain in a rat model of peripheral nerve injury. Anesthesiology 2004; 101: 476–487.
Wu ZZ, Guan BC, Li ZW, Yang Q, Liu CJ, Chen JG . Sustained potentiation by substance P of NMDA-activated current in rat primary sensory neurons. Brain Res 2004; 1010: 117–126.
Todorovic SM, Lingle CJ . Pharmacological properties of T-type Ca2+ current in adult rat sensory neurons: effects of anticonvulsant and anesthetic agents. J Neurophysiol 1998; 79: 240–252.
Acknowledgements
This study was funded in part by the VA Rehabilitation Research and Development grant 3690-03 to QHH.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Gene Therapy website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Fischer, G., Pan, B., Vilceanu, D. et al. Sustained relief of neuropathic pain by AAV-targeted expression of CBD3 peptide in rat dorsal root ganglion. Gene Ther 21, 44–51 (2014). https://doi.org/10.1038/gt.2013.56
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gt.2013.56
Keywords
This article is cited by
-
Gene therapy for chronic pain: emerging opportunities in target-rich peripheral nociceptors
Nature Reviews Neuroscience (2023)
-
Sigma-1 receptor activity in primary sensory neurons is a critical driver of neuropathic pain
Gene Therapy (2022)
-
Inhibition of autophagy by CRMP2-derived peptide ST2-104 (R9-CBD3) via a CaMKKβ/AMPK/mTOR pathway contributes to ischemic postconditioning-induced neuroprotection against cerebral ischemia-reperfusion injury
Molecular Brain (2021)
-
Nasal delivery of a CRMP2-derived CBD3 adenovirus improves cognitive function and pathology in APP/PS1 transgenic mice
Molecular Brain (2020)
-
AAV-encoded CaV2.2 peptide aptamer CBD3A6K for primary sensory neuron-targeted treatment of established neuropathic pain
Gene Therapy (2019)
