
Abstract
Alzheimer disease is the most common cause of dementia. It occurs worldwide and affects all ethnic groups. The incidence of Alzheimer disease is increasing due, in part, to increased life expectancy and the aging baby boomer generation. The average lifetime risk of developing Alzheimer disease is 10–12%. This risk at least doubles with the presence of a first-degree relative with the disorder. Despite its limited utility, patients express concern over their risk and, in some instances, request testing. Furthermore, research has demonstrated that testing individuals for apolipoprotein E can be valuable and safe in certain contexts. However, because of the complicated genetic nature of the disorder, few clinicians are prepared to address the genetic risks of Alzheimer disease with their patients. Given the increased awareness in family history thanks to family history campaigns, the increasing incidence of Alzheimer disease, and the availability of direct to consumer testing, patient requests for information is increasing. This practice guideline provides clinicians with a framework for assessing their patients' genetic risk for Alzheimer disease, identifying which individuals may benefit from genetic testing, and providing the key elements of genetic counseling for AD.
Similar content being viewed by others


Main
Alzheimer disease (AD) currently affects more than 5 million Americans.1 Although the majority of cases occur in the elderly, approximately 250,000 people have early-onset AD (EOAD) with onset of symptoms before age 65 years.1 Individuals with a family history of dementia often worry about their own risk of developing dementia and may be interested in learning about genetic testing options.2,3 Although genetic testing for AD has become more accessible through clinical laboratories and direct-to-consumer testing, the understanding of AD genetics continues to evolve, making it difficult for clinicians to effectively counsel their patients on hereditary risk and genetic testing.4 The purpose of this practice guideline is to provide clinicians with a framework for assessing their patients' genetic risk for AD, identifying which individuals may benefit from genetic testing, and providing the key elements of genetic counseling for AD, which is an integral part of the testing protocol.
CLINICAL ASPECTS OF AD
AD is the most common form of dementia, accounting for two thirds of all dementia diagnoses.5 It is usually marked by slowly progressive episodic memory loss, which evolves into global loss of cognitive ability, psychiatric features, and involvement of other deeper brain areas such as the basal ganglia.6 Atypical presentations occur in 6–14% of autopsy confirmed cases.7–10 As the disease progresses, motor dysfunction, such as parkinsonism, myoclonus, spastic paraparesis, and seizures, may also accompany neurologic and neuropsychiatric symptoms.7,11 The variability in phenotype increases the challenges of clinical management and appropriate use of genetic testing.12
Current diagnostic criteria for AD are based on the Diagnostic and Statistical Manual for Mental Disorders, 4th Edition, and the National Institute of Neurological Disorders, Communicative Disorders and Stroke-Alzheimer Disease and Related Disorders Association working group.13,14 A definitive diagnosis of AD can only be made by pathologic confirmation (National Institute on Aging-Reagan criteria) after autopsy of a symptomatic individual or identification of a pathogenic mutation in a causal AD gene (see discussion later) in a symptomatic individual.7,15,16 The risk for developing AD is associated most heavily with genetic factors and age, but sex, level of education, and history of head trauma, among others under investigation, may also be contributing factors.1,17–19 Members of the general population are at approximately 10–12% risk of developing AD in their lifetime. More precise risk estimates are only available for those with a family history consistent with autosomal dominant EOAD and those with a first-degree relative with AD.
Research on biomarkers for AD with the purpose of diagnosis, prediction of disease progression, presymptomatic predictive testing, and clinical trial use is proceeding around the world. Results indicate that cerebrospinal fluid (CSF) beta amyloid peptides (Aβ1-42), total tau, and phosphorylated tau levels have significant correlation with disease status, with evidence for decreased Aβ1-42 and increased total tau and phosphorylated tau in people with mild cognitive impairment (MCI) and AD, when compared with controls.20–22 Neuroimaging using magnetic resonance imaging, positron emission tomography, and Pittsburgh Compound B positron emission tomography improves the validity of the CSF markers.23 However, studies on these biomarkers did not use standardized assays and techniques, resulting in variability in measurements between different laboratories and within studies.22,24 Currently, CSF biomarkers are being used by some physicians in the differential diagnosis of dementia indicating the likelihood that a patient has AD rather than another form of dementia. Caution needs to be taken when interpreting these tests as results are not conclusive. Similarly, these test results cannot be used to predict the time of conversion from MCI to AD or as a definitive diagnostic test.22,25 Of note, studies indicate that apolipoprotein E (APOE) ε4 carriers with MCI have lower Aβ1-42 than noncarriers with MCI, indicating increased probability of conversion to AD compared with other APOE genotypes.22,26
At the current time, treatment of AD is focused on slowing the disease progression through two primary processes: cholinesterase inhibitors and N-methyl-d-aspartic acid receptor antagonists. Some studies suggest that a lifestyle promoting good cardiovascular health may be beneficial in reducing the risk or delaying the age of onset of AD.27,28 In addition, nonpharmacologic treatments (environment modification and caregiver training) can be effective in managing some of the behavior problems associated with AD, such as depression, agitation/aggression, wandering, and sleep disturbance.29
GENETICS OF AD
Alzheimer genetics is traditionally subdivided into early onset (EOAD) and late onset (LOAD). EOAD has an onset before age 60–65 years and accounts for 1–5% of all cases.30–32 LOAD has an onset after age 60–65 years and is the predominant form of AD. Additionally, family history may be consistent with autosomal dominant, familial, or sporadic AD.
Autosomal dominant
Families with autosomal dominant AD are characterized by disease that occurs in at least three individuals in two or more generations, with two of the individuals being first-degree relatives of the third (e.g., grandparent, parent, and child). This pattern can sometimes be masked by small families, premature death due to other causes, or, rarely, by incomplete penetrance. Autosomal dominant AD, which represents <5% of cases, is seen almost exclusively in EOAD families, thus the terms are often used interchangeably.30,31 It is important to note, however, that not all EOAD is inherited in an autosomal dominant pattern. Mutations in known causative genes in such families are identified approximately 40–80% of the time with variability likely due to inconsistency in criteria used to define autosomal dominant AD.31,33–35
Familial
Familial AD is characterized by disease that occurs in more than one individual, and at least two of the affected individuals are third-degree relatives or closer. Familial clustering represents approximately 15–25% of AD cases. Most often these are families with LOAD (15–25% of all LOAD cases), but familial clustering can be seen in approximately 47% of EOAD cases.31,36 These familial EOAD cases may represent hidden autosomal dominant AD due to small family size or cases of premature death.
Sporadic
Sporadic AD is characterized by an isolated case in the family or cases separated by more than three degrees of relationship. Sporadic AD represents approximately 75% of all cases. Typically, sporadic cases are LOAD, but approximately 40% of EOAD cases may be classified as sporadic possibly representing hidden familial or autosomal dominant disease, particularly if the family size is small and/or there are case(s) of premature death.31,37
Currently, there are three known deterministic (causative) genes in which mutations are associated with autosomal dominant EOAD: PSEN1, PSEN2, and APP (Table 1).31,33,38–54 The major pathologic processes in AD seem to be the formation of neurofibrillary tangles and the deposition of Αβ42 into senile plaques in the brain. Thus, it should not be surprising that each of the known AD genes play a role in the production, trafficking, and clearance of Αβ42.55 Overall, however, mutations in the EOAD genes are rare, and account for only 1–5% of all cases of AD.30,31 Because not all families with autosomal dominant EOAD have identifiable mutations in PSEN1, PSEN2, or APP, it is likely there are additional genes that influence the pathophysiology of EOAD.31,49 Predictive/presymptomatic testing for autosomal dominant EOAD is most informative when a mutation has been confirmed in a symptomatic family member. Mutations in PSEN1 and APP are associated with complete penetrance, meaning that all individuals who have a PSEN1 or APP mutation will develop AD if they live a normal lifespan. In contrast, mutations in PSEN2 show 95% penetrance, meaning that not everyone with a PSEN2 mutation will develop AD.41,53 In general, clear genotype-phenotype correlations cannot typically be made for the three causative genes, and age of onset can vary more than 20 years within the same family. Although several mutations in PSEN1, PSEN2, and APP have been reported to demonstrate a distinct clinical or neuropathological phenotype, care must be taken when interpreting test results, as mutations may have variable expression both within and between families.56–58 Two other complicating factors are the ambiguity of a negative result in the absence of a known familial mutation and identifying a variant of uncertain significance.59
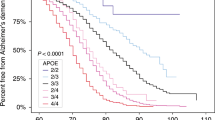
In contrast to these three deterministic genes, a fourth gene, APOE has been identified as a susceptibility gene for AD. APOE has three isoforms, ε2, ε3, and ε4. The ε4 variant of APOE is associated with an increased risk for AD, most notably in LOAD, but also EOAD.17,33,37,48,60,61 Approximately 50–70% of people with AD carry at least one ε4 allele.17,18,62,63 This percentage increases in those with AD who have a positive family history of disease.64 In addition, ε4 homozygotes are considerably more frequent among those with EOAD than LOAD.31,47,65 The presence of an ε4 allele increases the risk of AD in a dose-dependent fashion; individuals with two copies of the ε4 allele are at the highest risk. Estimates of the increased risk conferred by ε4 differ widely and also differ based on age and gender.31,66–68 Typical estimates suggest a 2–3-fold increased risk for ε4 heterozygotes, whereas estimates for homozygotes vary anywhere from a 2-fold to 10-fold increased risk.39,69–71 Many studies about the effects of the ε4 allele suggest that it influences the age at which AD occurs, rather than the overall lifetime risk for AD.17,61,72–75 Several studies have also suggested that the presence of an ε2 allele may play a protective role against developing AD.74,75 In addition to its role in AD, APOE has also been associated with risks of other diseases such as cardiovascular disease and macular degeneration.55,76 In the absence of APOE testing, first-degree relatives of an affected individual in sporadic or familial cases are estimated to have a 15–39% lifetime risk of developing AD, a 2–4-fold increase compared with the general population.77–83
PEDIATRIC TESTING FOR AD
Given the great variability in both symptoms and age of onset, even in family members who share the same causative mutation, genotypic information provides very little in the way of anticipatory guidance for the child. In addition, because AD remains, at this time, a disease that can neither be prevented nor cured, it is difficult to argue that there is a medical or social benefit to knowing the genotype of the child.84,85
GENETIC TESTING FOR AUTOSOMAL DOMINANT AD IN ADULTS
The use of genetic testing for diagnostic purposes in early-onset autosomal dominant AD has long been debated by clinicians in the dementia field.50,86,87 Although mutations are rare and testing may reveal variants of unknown significance, genetic testing may result in definitive diagnosis, improve understanding for the family, and allow at-risk relatives to have the option of predictive testing.88,89 Genetic counseling for symptomatic patients should be performed in the presence of the individual's legal guardian or family member to help assess the level to which he/she is able to understand the purpose and possible results of the genetic test and to provide informed consent (Fig. 1).90

Protocols for genetic testing for AD.
With respect to asymptomatic individuals, there are concerns genetic testing may trigger an untoward psychological response, such as severe depression, anxiety, or even suicidal ideation.91 However, research studies and clinical experience indicate that the majority of those tested using a standardized counseling protocol demonstrated effective coping skills and absence of negative psychological reactions after several months and found the testing to be beneficial, although the long-term effects of predictive testing for EOAD remain to be seen.92–95 Common reasons for testing included concern about early symptoms of dementia, financial or family planning, and relief from anxiety.92
The Huntington Disease Society of America's Guidelines for Genetic Testing for Huntington Disease (HD) is considered by many to be the gold standard for genetic testing for adult onset conditions.96,97 This guideline uses a multidisciplinary approach to facilitate autonomous decision making. It includes a two-part pretest and one or more posttest/disclosure genetic counseling sessions conducted in person, in the presence of a patient-selected support person who accompanies the patient to all genetic counseling visits. It also uses a neurologic evaluation and a psychiatric evaluation. Through this process, the various physical, psychological, social, and family history factors that influence the pretest decision-making process are brought to light, allowing the individual to make an informed decision about genetic testing while minimizing the risk of adverse outcome.
APOE SUSCEPTIBILITY TESTING
Although the association of the APOE ε4 allele with AD was identified in 1993, APOE genotyping for predictive purposes is not routinely offered in a clinical setting.98 Because the ε4 allele is neither necessary nor sufficient to cause AD, there have been numerous consensus statements and articles that have recommended against using APOE genotyping for predicting AD risk.6,15,16,99–102 The primary reasons for this recommendation include low sensitivity and specificity of testing, lack of preventive options, and the difficult nature of effectively conveying probabilistic risk.103 In addition, the association data indicate that risk conferred by APOE differs between genders and possibly by ethnic group.66–68 Because group differences in relative risk could result from differences in the prevalence of environmental exposures, modifier gene variants, or other factors (e.g., the effects of hormones in the case of gender differences), these differences complicate test result interpretation.
Finally, the risk associated with the ε4 allele not only varies widely between studies but also is often reported as an odds ratio, which is difficult to translate into meaningful figures for individual counseling.68,104–107 Cumulative risk curves for first-degree relatives of patients with AD stratified by APOE genotype and gender have been published but do not take into consideration multiple first- and second-degree relatives affected with AD and are based only on population studies of individuals of European or African American descent.106,108,109
A series of three successive multicenter, randomized, controlled trials, called the Risk Evaluation and Education for Alzheimer's Disease (REVEAL) study was initiated to evaluate the potential benefits and harms of APOE genotyping in the clinical context.108,110 Findings indicated that disclosing APOE genotype status to adult children of parents diagnosed with AD did not result in significant short-term psychological risk to this population.110 In addition, findings showed that the degree to which participants recalled the information they were given was a reflection on both the manner in which the risk was communicated and the complexity of the information itself, indicating the importance of good counseling.111 Approximately half of participants who correctly recalled their risk information actually believed their risk was significantly different than what was disclosed to them.112 This finding suggests that counselors should recognize a possible “anchoring and adjustment” bias whereby test recipients adhere to baseline risk perceptions even in the face of disconfirming evidence. The most common reasons for undergoing testing were desire for information, desire to feel in control, arranging personal care, advance planning, and emotional coping.113,114 Another finding was that participants with an ε4 allele were more likely than other groups to report health behavior changes, including the new use of dietary supplements.115 Although improvements in health behavior may be a beneficial outcome, there is concern such individuals will be vulnerable to marketing of treatments or supplements in the absence of evidence-based recommendations (e.g., direct to consumer [DTC] nutrigenomics testing).116 Another behavioral outcome is that participants receiving an ε4 result were significantly more likely than controls to report long-term care insurance changes during the 1-year follow-up period.117 From a policy perspective, these findings suggest the need to discuss issues of potential genetic discrimination in light of limitations to current state and federal genetic discrimination legislation.
It should be noted that the REVEAL study is not representative of a typical clinical setting. The cohort was a preselected (having a parent with AD), self-selecting (chose to participate), highly educated group. In addition, REVEAL participants obtained testing free of charge, and results were not included in their medical record.118
GUIDELINES
-
Pediatric testing for AD should not occur. Prenatal testing for AD is not advised if the patient intends to continue a pregnancy with a mutation.
-
Genetic testing for AD should only occur in the context of genetic counseling (in-person or through videoconference) and support by someone with expertise in this area.
-
Symptomatic patients: Genetic counseling for symptomatic patients should be performed in the presence of the individual's legal guardian or family member.
-
Asymptomatic patients: A protocol based on the International Huntington Association and World Federation of Neurology Research Group on Huntington's Chorea Guidelines is recommended.
-
DTC APOE testing is not advised.
-
A ≥3-generation family history should be obtained, with specific attention to the age of onset of any neurologic and/or psychiatric symptoms, type of dementia and method of diagnosis, current ages, or ages at death (especially unaffected relatives), and causes of death. Medical records should be used to confirm AD diagnosis when feasible. The history of additional relatives may prove useful, especially in small families or those with a preponderance of early death that may mask a history of dementia.
-
A risk assessment should be performed by pedigree analysis to determine whether the family history is consistent with EOAD or LOAD and with autosomal dominant (with or without complete penetrance), familial, or sporadic inheritance.
-
Patients should be informed that currently there are no proven pharmacologic or lifestyle choices that reduce the risk of developing AD or stop its progression.
-
The following potential genetic contributions to AD should be reviewed:
-
○ The lifetime risk of AD in the general population is approximately 10–12% in a 75–80-year lifespan.
-
○ The effect(s) of ethnicity on risk is still unclear.
-
○ Although some genes are known, there are very likely others (susceptibility, deterministic, and protective) whose presence and effects are currently unknown.
For families in which an autosomal dominant AD gene mutation is a possibility
-
Discuss the risk of inheriting a mutation from a parent affected with autosomal dominant AD is 50%. In the absence of identifying a mutation in apparent autosomal dominant families, risk to offspring could be as high as 50% but may be less.
-
Testing for genes associated with early-onset autosomal dominant AD should be offered in the following situations:
-
○ A symptomatic individual with EOAD in the setting of a family history of dementia or in the setting of an unknown family history (e.g., adoption).
-
○ Autosomal dominant family history of dementia with one or more cases of EOAD.
-
○ A relative with a mutation consistent with EOAD (currently PSEN1/2 or APP).
The Alzheimer Disease & Frontotemporal Dementia Mutation Database should be consulted (www.molgen.ua.ac.be/ADMutations/) before disclosure of genetic test results, and specific genotypes should not be used to predict the phenotype in diagnostic or predictive testing (Table 2).
-
Discuss the likelihood of identifying a mutation in PSEN1, PSEN2, or APP, noting that current experience indicates that this likelihood decreases with lower proportions of affected family members and/or older ages of onset.
-
Ideally, an affected family member should be tested first. If no affected family member is available for testing and an asymptomatic individual remains interested in testing despite counseling about the low likelihood of an informative result (a positive result for a pathogenic mutation), he/she should be counseled according to the recommended protocol. If the affected relative, or their next of kin, is uninterested in pursuing tested, the option of DNA banking should be discussed.
For families in which autosomal dominant AD is unlikely
-
Inform them why their family history is consistent with familial or sporadic AD.
-
Discuss that both sporadic and familial cases can be due to a genetic susceptibility. Risk estimates are only available for first-degree relatives of an affected individual in sporadic or familial cases.
-
Genetic testing for susceptibility loci (e.g., APOE) is not clinically recommended due to limited clinical utility and poor predictive value. If a patient wishes to pursue testing despite genetic counseling and recommendations to the contrary, testing may be considered at the clinician's discretion. Testing performed should follow the HD genetic testing guidelines, with emphasis on genetic counseling with a qualified clinician. As such, DTC genetic testing is not advised.
-
Motives and considerations for pursuing genetic testing should be explored. This counseling should be an exploration of personal experiences, value and beliefs, and personal and family needs. Genetic testing should be discussed within the context of adapting to familial risk and when clients feel compelled to learn a more refined estimate of their risks to enhance their quality of life. As part of this, it is helpful to lead the individual through the scenario of receiving a positive test result and a negative test result, having them assess the ways these results would positively or adversely impact their psyche, life plans, and relationships.
-
○ Symptomatic patients: Because genetic testing of a symptomatic individual is typically requested by a relative concerned about his risk, the counselor must remain alert to any potential conflicts of interest, such as lack of interest of the symptomatic patient or of other at-risk family members. If the symptomatic patient gives any inclination of being averse to testing, it is not recommended. Instead, DNA banking should be explored.
-
If there is disagreement within the family regarding whether testing should be performed, a family meeting is strongly encouraged (with or without the genetic counselor present). A family meeting would allow all interested parties to discuss the potential impact of the genetic testing on the family, how test results will be communicated, and how to respect the rights of those family members who do not wish to know the results.
-
Genetic testing: In the event testing is chosen, the following is recommended:
-
○ Asymptomatic patients should receive a neurologic examination to assess for signs of dementia and to establish a baseline.
-
○ Assess patient's and any accompanying family member's psychological state of mind. In the case of presymptomatic testing, a consultation with a psychologist/psychiatrist may be recommended for the patient as part of the HD testing approach.
-
If the patient seems to suffer from, or is potentially at risk for significant psychological/psychiatric problems, consider a psychotherapy referral before testing.
-
If the psychological assessment suggests testing is not in the person's current best interest (e.g., untreated depression or recent death), these reservations should be shared openly, and an agreement should be made to revisit testing once the underlying condition and/or stressors have diminished. A referral for psychotherapy may also be appropriate.
-
○ Assess and review the psychosocial impact of testing on the patient and his/her family.
-
Reinforce results cannot be “taken back” (although an individual can decide not to learn his or her test results after having the test performed.)
-
○ Discuss testing logistics, associated costs, and possible outcomes.
-
For EOAD genes, determine best approach to testing for patient (i.e., stepwise testing beginning with PSEN1 as the most likely gene or ordering a panel).
-
Discuss where results will be kept (e.g., medical record).
-
Determine who will accompany the patient to the result session for support.
-
Discuss possible test outcomes (positive, negative, or variant of uncertain significance). If testing for APOE, consider whether you will report other disease risk implications. If so, these should be included in the discussion of test outcomes with the patient. Also, it should be reiterated that APOE is a susceptibility gene and is not a predictive test. Thus, individuals with no copies of the ε4 allele still face a 2–4-fold increased lifetime risk of developing AD if they have a first-degree relative with AD.
-
○ Assist the patient and participating family members with informed decision making regarding whom, if anyone, they plan to share the results with and how. Inform about the importance of discretion when discussing genetic testing and results.
-
○ Discuss the potential impact of genetic test results on insurance, and the benefits and limitations of existing state and federal genetic discrimination legislation.
-
○ Obtain informed consent for all genetic testing for AD.
-
○ After results disclosure, revisit the individual's plans regarding with whom and how the results will be shared.
-
○ Arrange for a follow-up plan to “check in” with the patient and, if relevant, participating family member, and determine whether another genetic counseling session would be beneficial to the patient and/or the patient's partner/family members/friends.
-
Discuss the availability and status of AD research and/or DNA banking.
References
Alzheimer's Association 2001 Alzheimer's disease facts and figures. Alzheimer's & Dementia 2001; 27: 208–244.
Neumann PJ, Hammitt JK, Mueller C, et al. Public attitudes about genetic testing for Alzheimer's disease. Health Aff (Millwood) 2001; 20: 252–264.
Roberts JS . Anticipating response to predictive genetic testing for Alzheimer's disease: a survey of first-degree relatives. Gerontologist 2000; 40: 43–52.
Chase GA, Geller G, Havstad SL, Holtzman NA, Bassett SS . Physicians' propensity to offer genetic testing for Alzheimer's disease: results from a survey. Genet Med 2002; 4: 297–303.
Nussbaum RL, Ellis CE . Alzheimer's disease and Parkinson's disease. N Engl J Med 2003; 348: 1356–1364.
Small GW, Rabins PV, Barry PP, et al. Diagnosis and treatment of Alzheimer disease and related disorders. Consensus statement of the American Association for Geriatric Psychiatry, the Alzheimer's Association, and the American Geriatrics Society. JAMA 1997; 278: 1363–1371.
Galton CJ, Patterson K, Xuereb JH, Hodges JR . Atypical and typical presentations of Alzheimer's disease: a clinical, neuropsychological, neuroimaging and pathological study of 13 cases. Brain 2000; 123: 484–498.
Johnson JK, Head E, Kim R, Starr A, Cotman CW . Clinical and pathological evidence for a frontal variant of Alzheimer disease. Arch Neurol 1999; 56: 1233–1239.
Larner AJ . “Frontal variant Alzheimer's disease”: a reappraisal. Clin Neurol Neurosurg 2006; 108: 705–708.
Kramer JH, Miller BL . Alzheimer's disease and its focal variants. Semin Neurol 2000; 20: 447–454.
Scarmeas N, Hadjigeorgiou GM, Papadimitriou A, et al. Motor signs during the course of Alzheimer disease. Neurology 2004; 63: 975–982.
Langa KM, Foster NL, Larson EB . Mixed dementia: emerging concepts and therapeutic implications. JAMA 2004; 292: 2901–2908.
American Psychiatric Association Diagnostic and statistical manual of mental disorders, 4th ed. Washington, DC, American Psychiatric Association, 1994.
McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM . Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984; 34: 939–944.
Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Neurobiol Aging 1997; 18: S1–S2.
Waldemar G, Dubois B, Emre M, et al. Recommendations for the diagnosis and management of Alzheimer's disease and other disorders associated with dementia: EFNS guideline. Eur J Neurol 2007; 14: e1–e26.
Breitner JC, Wyse BW, Anthony JC, et al. APOE-epsilon4 count predicts age when prevalence of AD increases, then declines: the Cache County Study. Neurology 1999; 53: 321–331.
Jarvik L, LaRue A, Blacker D, et al. Children of persons with Alzheimer disease: what does the future hold?. Alzheimer Dis Assoc Disord 2008; 22: 6–20.
Coats M, Morris JC . Antecedent biomarkers of Alzheimer's disease: the adult children study. J Geriatr Psychiatry Neurol 2005; 18: 242–244.
Shaw LM, Vanderstichele H, Knapik-Czajka M, et al. Alzheimer's disease neuroimaging initiative. Cerebrospinal fluid biomarker signature in Alzheimer's disease: neuroimaging initiative subjects. Ann Neurol 2009; 65: 403–413.
Sunderland T, Linker G, Mirza N, et al. Decreased β-amyloid1–42 and increased tau levels in cerebrospinal fluid of patients with Alzheimer disease. JAMA 2003; 289: 2094–2103.
Mattsson N, Zetterberg H, Hansson O, et al. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA 2009; 302: 385–393.
Vemuri P, Wiste HJ, Weigand SD, et al. Alzheimer's disease neuroimaging initiative. Serial MRI and CSF biomarkers in normal aging, MCI, and AD. Neurology 2010; 75: 143–151.
Verwey NA, van der Flier WM, Blennow K, et al. A worldwide multicentre comparison of assays for cerebrospinal fluid biomarkers in Alzheimer's disease. Ann Clin Biochem 2009; 46: 235–240.
Petersen RC, Trojanowski JQ . Use of Alzheimer disease biomarkers: potentially yes for clinical trials but not yet for clinical practice. JAMA 2009; 302: 436–437.
Vemuri P, Wiste HJ, Weigand SD, et al. Alzheimer's disease neuroimaging initiative. Effect of apolipoprotein E on biomarkers of amyloid load and neuronal pathology in Alzheimer disease. Ann Neurol 2010; 67: 308–316.
Larson EB, Wang L, Bowen JD, et al. Exercise is associated with reduced risk for incident dementia among persons 65 years of age and older. Ann Intern Med 2006; 144: 73–81.
Solfrizzi V, Capurso C, D'Introno A, et al. Lifestyle-related factors in predementia and dementia syndromes. Expert Rev Neurother 2008; 8: 133–158.
Teri L, Logsdon RG, McCurry SM . Nonpharmacologic treatment of behavioral disturbance in dementia. Med Clin North Am 2002; 86: 641–656, viii
Bertram L, Tanzi RE . The genetic epidemiology of neurodegenerative disease. J Clin Invest 2005; 115: 1449–1457.
Campion D, Dumanchin C, Hannequin D, et al. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet 1999; 65: 664–670.
Alzheimer's Association. Early onset dementia: a national challenge, a future-crisis. Washington, DC: Alzheimer's Association, 2006. Available at: http://www.alz.org. Accessed June 2006.
Janssen JC, Beck JA, Campbell TA, et al. Early onset familial Alzheimer's disease: mutation frequency in 31 families. Neurology 2003; 60: 235–239.
Kowalska A, Wender M, Florczak J, et al. Molecular genetics of Alzheimer's disease: presenilin 1 gene analysis in a cohort of patients from the Poznan region. J Appl Genet 2003; 44: 231–234.
Tedde A, Forleo P, Nacmias B, et al. A presenilin-1 mutation (Leu392Pro) in a familial AD kindred with psychiatric symptoms at onset. Neurology 2000; 55: 1590–1591.
Brickell KL, Steinbart EJ, Rumbaugh M, et al. Early-onset Alzheimer disease in families with late-onset Alzheimer disease: a potential important subtype of familial Alzheimer disease. Arch Neurol 2006; 63: 1307–1311.
Van Duijn CM, de Knijiff P, Cruts M, et al. Apolipoprotein E4 allele in a population-based study of early-onset Alzheimer's disease. Nat Genet 1994; 7: 74–78.
Levy-Lahad E, Wasco W, Poorkaj P, et al. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science 1995; 269: 973–977.
Frikke-Schmidt R, Nordestgaard BG, Thudium D, Moes Gronholdt ML, Tybjaerg-Hansen A . APOE genotype predicts AD and other dementia but not ischemic cerebrovascular disease. Neurology 2001; 56: 194–200.
Goate A, Chartier-Harlin MC, Mullan M, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature 1991; 349: 704–706.
Sherrington R, Froelich S, Sorbi S, et al. Alzheimer's disease associated with mutations in presenilin 2 is rare and variably penetrant. Hum Mol Genet 1996; 5: 985–988.
Tanzi RE, Vaula G, Romano DM, et al. Assessment of amyloid beta-protein precursor gene mutations in a large set of familial and sporadic Alzheimer disease cases. Am J Hum Genet 1992; 51: 273–282.
Schellenberg GD, Bird TD, Wijsman EM, et al. Genetic linkage evidence for a familial Alzheimer's disease locus on chromosome 14. Science 1992; 258: 668–671.
Van Broeckhoven C, Backhovens H, Cruts M, et al. Mapping of a gene predisposing to early-onset Alzheimer's disease to chromosome 14q24.3. Nat Genet 1992; 2: 335–339.
St George-Hyslop P, Haines J, Rogaev E, et al. Genetic evidence for a novel familial Alzheimer's disease locus on chromosome 14. Nat Genet 1992; 2: 330–334.
Rogaev EI, Sherrington R, Rogaeva EA, et al. Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature 1995; 376: 775–778.
Cruts M, Van Broeckhoven C . Presenilin mutations in Alzheimer's disease. Hum Mutat 1998; 11: 183–190.
Cruts M, van Duijin CM, Backhovens H, et al. Estimation of the genetic contribution of presenilin-1 and -2 mutations in a population-based study of presenile Alzheimer disease. Hum Mol Genet 1998; 7: 43–51.
Raux G, Guyant-Marechal L, Martin C, et al. Molecular diagnosis of autosomal dominant early onset Alzheimer's disease: an update. J Med Genet 2005; 42: 793–795.
Finckh U, Alberici A, Antoniazzi M, et al. Variable expression of familial Alzheimer disease associated with presenilin 2 mutation M239I. Neurology 2000; 54: 2006–2008.
Bird TD, Levy-Lahad E, Poorkaj P, et al. Wide range in age of onset for chromosome 1-related familial Alzheimer's disease. Ann Neurol 1996; 40: 932–936.
Rogaeva EA, Fafel KC, Song YQ, et al. Screening for PS1 mutations in a referral-based series of AD cases: 21 novel mutations. Neurology 2001; 57: 621–625.
Tedde A, Nacmias B, Ciantelli M, et al. Identification of new presenilin gene mutations in early-onset familial Alzheimer disease. Arch Neurol 2003; 60: 1541–1544.
Lleo A, Blesa R, Queralt R, et al. Frequency of mutations in the presenilin and amyloid precursor protein genes in early-onset Alzheimer disease in Spain. Arch Neurol 2002; 59: 1759–1763.
Tabaton M, Tamagno E . The molecular link between beta- and gamma-secretase activity on the amyloid beta precursor protein. Cell Mol Life Sci 2007; 64: 2211–2218.
Ataka S, Tomiyama T, Takuma H, et al. A novel presenilin-1 mutation (Leu85Pro) in early-onset Alzheimer disease with spastic paraparesis. Arch Neurol 2004; 61: 1773–1776.
Mann DM, Pickering-Brown SM, Takeuchi A, Iwatsubo T . Amyloid angiopathy and variability in amyloid beta deposition is determined by mutation position in presenilin-1-linked Alzheimer's disease. Am J Pathol 2001; 158: 2165–2175.
Athan ES, Williamson J, Ciappa A, et al. A founder mutation in presenilin 1 causing early-onset Alzheimer disease in unrelated Caribbean Hispanic families. JAMA 2001; 286: 2257–2263.
Dermaut B, Cruts M, Slooter AJ, et al. The Glu318Gly substitution in presenilin 1 is not causally related to Alzheimer disease. Am J Hum Genet 1999; 64: 290–292.
Pericak-Vance MA, Bebout JL, Gaskell PC Jr, et al. Linkage studies in familial Alzheimer disease: evidence for chromosome 19 linkage. Am J Hum Genet 1991; 48: 1034–1050.
Khachaturian AS, Corcoran CD, Mayer LS, Zandi PP, Breitner JC . Apolipoprotein E epsilon4 count affects age at onset of Alzheimer disease, but not lifetime susceptibility: the Cache County Study. Arch Gen Psychiatry 2004; 61: 518–524.
Ashford JW, Mortimer JA . Non-familial Alzheimer's disease is mainly due to genetic factors. J Alzheimers Dis 2002; 4: 169–177.
National Institute on Aging Progress report on AD. Rockville, MD, National Institutes of Health, 1995.
Jarvik G, Larson EB, Goddard K, Schellenberg GD, Wijsman EM . Influence of apoliprotein E genotype on the transmission of Alzheimer disease in a community-based sample. Am J Hum Genet 1996; 58: 191–200.
Zuo L, van Dyck CH, Luo X, Kranzler HR, Yang BZ, Gelernter J . Variation at APOE and STH loci and Alzheimer's disease. Behav Brain Funct 2006; 2: 13.
Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997; 278: 1349–1356.
Devi G, Ottman R, Tang M, et al. Influence of APOE genotype on familial aggregation of AD in an urban population. Neurology 1999; 53: 789–794.
Martinez M, Campion D, Brice A, et al. Apolipoprotein E epsilon4 allele and familial aggregation of Alzheimer disease. Arch Neurol 1998; 55: 810–816.
Qiu C, Kivipelto M, Aguero-Torres H, Winblad B, Fratiglioni L . Risk and protective effects of the APOE gene towards Alzheimer's disease in the Kungsholmen project: variation by age and sex. J Neurol Neurosurg Psychiatry 2004; 75: 828–833.
Slooter AJ, Cruts M, Kalmijn S, et al. Risk estimates of dementia by apolipoprotein E genotypes from a population-based incidence study: the Rotterdam Study. Arch Neurol 1998; 55: 964–968.
Plassman BL, Langa KM, Fisher GG, et al. Prevalence of dementia in the United States: the aging, demographics, and memory study. Neuroepidemiology 2007; 29: 125–132.
Meyer MR, Tschanz JT, Norton MC, et al. APOE genotype predicts when–not whether–one is predisposed to develop Alzheimer disease. Nat Genet 1998; 19: 321–322.
Olarte L, Schupf N, Lee JH, et al. Apolipoprotein E epsilon4 and age at onset of sporadic and familial Alzheimer disease in Caribbean Hispanics. Arch Neurol 2006; 63: 1586–1590.
Corder EH, Saunders AM, Risch NJ, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet 1994; 7: 180–184.
Saunders AM . Apolipoprotein E and Alzheimer disease: an update on genetic and functional analyses. J Neuropathol Exp Neurol 2000; 59: 751–758.
Bennet AM, Di Angelantonio E, Ye Z, et al. Association of apolipoprotein E genotypes with lipid levels and coronary risk. JAMA 2007; 298: 1300–1311.
Korten AE, Jorm AF, Henderson AS, Broe GA, Creasey H, McCusker E . Assessing the risk of Alzheimer's disease in first-degree relatives of Alzheimer's disease cases. Psychol Med 1993; 23: 915–923.
Silverman JM, Raiford K, Edland S, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part VI. Family history assessment: a multicenter study of first-degree relatives of Alzheimer's disease probands and nondemented spouse controls. Neurology 1994; 44: 1253–1259.
Seshadri S, Drachman DA, Lippa CF . Apolipoprotein E epsilon 4 allele and the lifetime risk of Alzheimer's disease. What physicians know, and what they should know. Arch Neurol 1995; 52: 1074–1079.
Liddell MB, Lovestone S, Owen MJ . Genetic risk of Alzheimer's disease: advising relatives. Br J Psychiatry 2001; 178: 7–11.
Lautenschlager NT, Cupples LA, Rao VS, et al. Risk of dementia among relatives of Alzheimer's disease patients in the MIRAGE study: what is in store for the oldest old?. Neurology 1996; 46: 641–650.
Farrer LA, O'Sullivan DM, Cupples LA, Growdon JH, Myers RH . Assessment of genetic risk for Alzheimer's disease among first-degree relatives. Ann Neurol 1989; 25: 485–493.
Farrer LA, Cupples LA, van Duijn CM, et al. Apoliprotein E genotype in patients with Alzheimer disease: implications for the risk of dementia among relatives. Ann Neurol 1995; 38: 797–808.
Points to consider: ethical, legal, and psychosocial implications of genetic testing in children and adolescents. American Society of Human Genetics Board of Directors, American College of Medical Genetics Board of Directors. Am J Hum Genet 1995; 57: 1233–1241.
Nelson RM, Botkin JR, Kodish ED, et al. Ethical issues with genetic testing in pediatrics. Pediatrics 2001; 107: 1451–1455.
Croes EA, Dermaut B, van Der Cammen TJ, van Broeckhoven C, van Duijn CM . Genetic testing should not be advocated as a diagnostic tool in familial forms of dementia. Am J Hum Genet 2000; 67: 1033–1035.
van der Cammen TJ, Croes EA, Dermaut B, et al. Genetic testing has no place as a routine diagnostic test in sporadic and familial cases of Alzheimer's disease. J Am Geriatr Soc 2004; 52: 2110–2113.
Serretti A, Olgiati P, De Ronchi D . Genetics of Alzheimer's disease. A rapidly evolving field. J Alzheimers Dis 2007; 12: 73–92.
Williamson J, LaRusse S . Genetics and genetic counseling: recommendations for Alzheimer's disease, frontotemporal dementia, and Creutzfeldt-Jakob disease. Curr Neurol Neurosci Rep 2004; 4: 351–357.
Eaton ML . Surrogate decision making for genetic testing for Alzheimer disease. Genet Test 1999; 3: 93–97.
Tibben A, Stevens M, de Wert GM, Niermeijer MF, van Duijn CM, van Swieten JC . Preparing for presymptomatic DNA testing for early onset Alzheimer's disease/cerebral haemorrhage and hereditary Pick disease. J Med Genet 1997; 34: 63–72.
Steinbart EJ, Smith CO, Poorkaj P, Bird TD . Impact of DNA testing for early-onset familial Alzheimer disease and frontotemporal dementia. Arch Neurol 2001; 58: 1828–1831.
Molinuevo JL, Pintor L, Peri JM, et al. Emotional reactions to predictive testing in Alzheimer's disease and other inherited dementias. Am J Alzheimers Dis Other Demen 2005; 20: 233.
Lannfelt L, Axelman K, Lilius L, Basun H . Genetic counseling in a Swedish Alzheimer family with amyloid precursor protein mutation. Am J Hum Genet 1995; 56: 332–335.
Anonymous. Commentary: a patient's perspective on predictive testing for Alzheimer disease. J Genet Counsel 2000; 9: 343–344.
The Huntington's Disease Society of America Guidelines for genetic testing. New York, The Huntington's Disease Society of America, 1994.
Guidelines for the molecular genetics predictive test in Huntington's disease. International Huntington Association (IHA) and the World Federation of Neurology (WFN) Research Group on Huntington's Chorea. Neurology 1994; 44: 1533–1536.
Strittmatter WJ, Saunders AM, Schmechel D, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA 1993; 90: 1977–1981.
Statement on use of apolipoprotein E testing for Alzheimer disease. American College of Medical Genetics/American Society of Human Genetics Working Group on ApoE and Alzheimer disease. JAMA 1995; 274: 1627–1629.
Post SG, Whitehouse PJ, Binstock RH, et al. The clinical introduction of genetic testing for Alzheimer disease. An ethical perspective. JAMA 1997; 277: 832–836.
McConnell LM, Koenig BA, Greely HT, Raffin TA . Genetic testing and Alzheimer disease: has the time come? Alzheimer Disease Working Group of the Stanford Program in Genomics, Ethics & Society. Nat Med 1998; 4: 757–759.
Burke W, Pinsky LE, Press NA . Categorizing genetic tests to identify their ethical, legal, and social implications. Am J Med Genet 2001; 106: 233–240.
Mayeux R, Saunders AM, Shea S, et al. Utility of the apolipoprotein E genotype in the diagnosis of Alzheimer's disease. Alzheimer's Disease Centers Consortium on Apolipoprotein E and Alzheimer's Disease. N Engl J Med 1998; 338: 506–511.
Tang MX, Maestre G, Tsai WY, et al. Relative risk of Alzheimer disease and age-at-onset distributions, based on APOE genotypes among elderly African Americans, Caucasians, and Hispanics in New York City. Am J Hum Genet 1996; 58: 574–584.
Maestre G, Ottman R, Stern Y, et al. Apolipoprotein E and Alzheimer's disease: ethnic variation in genotypic risks. Ann Neurol 1995; 37: 254–259.
Cupples LA, Farrer LA, Sadovnick AD, Relkin N, Whitehouse P, Green RC . Estimating risk curves for first-degree relatives of patients with Alzheimer's disease: the REVEAL study. Genet Med 2004; 6: 192–196.
Li G, Silverman JM, Altstiel LD, et al. Apolipoprotein E-epsilon 4 allele and familial risk in Alzheimer's disease. Genet Epidemiol 1996; 13: 285–298.
Roberts JS, Cupples LA, Relkin NR, Whitehouse PJ, Green RC . Genetic risk assessment for adult children of people with Alzheimer's disease: the Risk Evaluation and Education for Alzheimer's Disease (REVEAL) study. J Geriatr Psychiatry Neurol 2005; 18: 250–255.
Christensen KD, Roberts JS, Royal CD, et al. Incorporating ethnicity into genetic risk assessment for Alzheimer disease: the REVEAL study experience. Genet Med 2008; 10: 207–214.
Green RC, Roberts JS, Cupples LA, et al. Disclosure of APOE genotype for risk of Alzheimer's disease. N Engl J Med 2009; 361: 11–20.
Eckert SL, Katzen H, Roberts JS, et al. Recall of disclosed apolipoprotein E genotype and lifetime risk estimate for Alzheimer's disease: the REVEAL Study. Genet Med 2006; 8: 746–751.
Linnenbringer E, Roberts JS, Hiraki S, Cupples LA, Green RC . “I know what you told me, but this is what I think”: perceived risk of Alzheimer's disease among individuals who accurately recall their genetics-based risk estimate. Genet Med 2010; 12: 219–227.
Roberts JS, LaRusse SA, Katzen H, et al. Reasons for seeking genetic susceptibility testing among first-degree relatives of people with Alzheimer disease. Alzheimer Dis Assoc Disord 2003; 17: 86–93.
Gooding HC, Linnenbringer EL, Burack J, Roberts JS, Green RC, Biesecker BB . Genetic susceptibility testing for Alzheimer disease: motivation to obtain information and control as precursors to coping with increased risk. Patient Educ Couns 2006; 64: 259–267.
Chao S, Roberts JS, Marteau TM, Silliman R, Cupples LA, Green RC . Health behavior changes after genetic risk assessment for Alzheimer's disease: the REVEAL study. Alzheimer Dis Assoc Disord 2008; 22: 94–97.
Kutz G Oral testimony before the Special Committee on Aging, United States Senate, Government Accountability Office. “NUTRIGENETIC TESTING”: tests purchased from four web sites mislead consumers. (Statement of Gregory Kutz, Managing Director Forensic Audits and Special Investigations). Available at: http://www.gao.gov/new.items/d06977t.pdf. Accessed July 27, 2006.
Zick CD, Mathews CJ, Roberts JS, Cook-Deegan R, Pokorski RJ, Green RC . Genetic testing for Alzheimer's disease and its impact on insurance purchasing behavior. Health Aff (Millwood) 2005; 24: 483–490.
Roberts JS, Barber M, Brown TM, et al. Who seeks genetic susceptibility testing for Alzheimer's disease? Findings from a multisite, randomized clinical trial. Genet Med 2004; 6: 197–203.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: The authors declare no conflict of interest.
Disclaimer: © 2011 National Society of Genetic Counselors and American College of Medical Genetics. All rights reserved. This document may not, in whole or in part, be reproduced, copied or disseminated, entered into or stored in a computer database or retrieval system, or otherwise utilized without the prior written consent of both the National Society of Genetic Counselors and American College of Medical Genetics.
The practice guidelines of the National Society of Genetic Counselors (NSGC) and the American College of Medical Genetics (ACMG) are developed by members of the NSGC and ACMG to assist geneticists, genetic counselors, and other health care providers in making decisions about appropriate management of genetic concerns; including access to and/or delivery of services. Each practice guideline focuses on a clinical or practice-based issue, and is the result of a review and analysis of current professional literature believed to be reliable. As such, information and recommendations within the practice guidelines reflect the current scientific and clinical knowledge at the time of publication, are only current as of their publication date, and are subject to change without notice as advances emerge.
In addition, variations in practice, which take into account the needs of the individual patient and the resources and limitations unique to the institution or type of practice, may warrant approaches, treatments and/or procedures that differ from the recommendations outlined in this guideline. Therefore, these recommendations should not be construed as dictating an exclusive course of management, nor does the use of such recommendations guarantee a particular outcome. Genetic counseling practice guidelines are never intended to displace a health care provider's best medical judgment based on the clinical circumstances of a particular patient or patient population. Practice guidelines are published by NSGC and ACMG for educational and informational purposes only, and NSGC and ACMG do not “approve” or “endorse” any specific methods, practices, or sources of information. It also would be prudent to consider whether intellectual property interests may restrict the performance of certain tests and other procedures.
Rights and permissions
About this article
Cite this article
Goldman, J., Hahn, S., Catania, J. et al. Genetic counseling and testing for Alzheimer disease: Joint practice guidelines of the American College of Medical Genetics and the National Society of Genetic Counselors. Genet Med 13, 597–605 (2011). https://doi.org/10.1097/GIM.0b013e31821d69b8
Published:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e31821d69b8
Keywords
This article is cited by
-
Shared and unique effects of ApoEε4 and pathogenic gene mutation on cognition and imaging in preclinical familial Alzheimer’s disease
Alzheimer's Research & Therapy (2023)
-
Superoxide dismutase: a key target for the neuroprotective effects of curcumin
Molecular and Cellular Biochemistry (2023)
-
The Role of Genetic Testing for Parkinson’s Disease
Current Neurology and Neuroscience Reports (2021)
-
Genetic counselling and testing for inherited dementia: single-centre evaluation of the consensus Italian DIAfN protocol
Alzheimer's Research & Therapy (2020)
-
Effects of gene mutation and disease progression on representative neural circuits in familial Alzheimer’s disease
Alzheimer's Research & Therapy (2020)
