
Abstract
Abstract: von Willebrand disease is a common inherited bleeding disorder characterized by excessive mucocutaneous bleeding. Characteristic bleeding symptoms include epistaxis, easy bruising, oral cavity bleeding, menorrhagia, bleeding after dental extraction, surgery, and/or childbirth, and in severe cases, bleeding into joints and soft tissues. There are three subtypes: types 1 and 3 represent quantitative variants and type 2 is a group of four qualitative variants: (1) type 2A—characterized by defective von Willebrand factor-dependent platelet adhesion because of decreased high-molecular-weight von Willebrand factor multimers, (2) type 2B—caused by pathologically increased von Willebrand factor-platelet interactions, (3) type 2M—caused by decreased von Willebrand factor-platelet interactions not based on the loss of high-molecular-weight multimers, and (4) type 2N—characterized by reduced binding of von Willebrand factor to factor VIII. The diagnosis of von Willebrand disease requires specialized assays of von Willebrand factor and/or molecular genetic testing of von Willebrand factor. Severe bleeding episodes can be prevented or controlled with intravenous infusions of virally inactivated plasma-derived clotting factor concentrates containing both von Willebrand factor and factor VIII. Depending on the von Willebrand disease type, mild bleeding episodes usually respond to intravenous or subcutaneous treatment with desmopressin, a vasopressin analog. Other treatments that can reduce symptoms include fibrinolytic inhibitors and hormones for menorrhagia.
Similar content being viewed by others
DIAGNOSIS
Clinical diagnosis
VWD is caused by deficient or defective plasma VWF, a large multimeric glycoprotein that plays a pivotal role in hemostasis by mediating platelet hemostatic function and stabilizing blood coagulation FVIII. It affects 0.16 to 1% of the population7,8; affected individuals registered at a tertiary care center number up to 1 in 10,000.9 The current International Society on Thrombosis and Hemostasis Scientific and Standardization Committee on VWF (ISTH SSC on VWF) VWD classification comprises three subtypes.10 Type 1 VWD is a partial quantitative deficiency of essentially normal VWF. Type 2 VWD is characterized by a qualitative deficiency and defective VWF (further subdivided into types 2A, 2B, 2M, and 2N). Type 3 VWD is a virtually complete quantitative deficiency of VWF.
VWD should be suspected in persons with excessive mucocutaneous bleeding, such as bruising without recognized trauma, prolonged, recurrent nose bleeds, and oral cavity bleeding, including bleeding from the gums after brushing or flossing teeth or prolonged bleeding after dental cleaning or extractions. Prolonged or excessive bleeding after surgery or trauma is often reported. Affected females also frequently experience menorrhagia (usually occurring since menarche)11 and prolonged or excessive bleeding after childbirth.12 The utility of standard clinical assessment tools to score occurrence of symptoms and their severity as part of VWD diagnosis is becoming recognized.13–15 These can determine whether there is more bleeding than in the normal population, justifying diagnosis of a bleeding disorder, and can help quantify extent of symptoms, indicating situations requiring clinical intervention.
Laboratory testing
Screening tests for VWD include a complete blood count, activated partial thromboplastin time, and prothrombin time.16,17 The complete blood count not only might be normal but could also show a microcytic anemia (if the individual is iron deficient) or a low platelet count (thrombocytopenia), specifically in type 2B VWD.18 The activated partial thromboplastin time is often normal but may be prolonged when the FVIII level is reduced to below 30–40 IU/dL (normal range is approximately 50–150 IU/dL) as can be seen in severe type 1 VWD, type 2N VWD, or type 3 VWD. The prothrombin time is normal in VWD. Although some laboratories may also include a skin bleeding time and platelet function analysis (closure time) in their evaluation of an individual with suspected VWD, these tests lack sensitivity in persons with mild bleeding.19–21
Hemostasis factor assays
The following specific hemostasis factor assays (Table 1) should be performed even if the screening tests are normal.22
VWF:Ag.
Quantity of VWF protein (antigen) in the plasma, measured antigenically using enzyme-linked immunosorbent assay or latex immunoassay. The normal range that should be determined independently by each laboratory is approximately 50–200 IU/dL.
VWF:RCo.
The ristocetin cofactor activity assay determines the ability of VWF to agglutinate platelets, initiated by the antibiotic ristocetin. The normal range is approximately 50–200 IU/dL.
FVIII:C level.
Functional FVIII assay that determines the activity of FVIII in the coagulation cascade. The normal range is approximately 50–150 IU/dL.
If abnormalities in the three tests above are identified, specialized coagulation laboratories may also perform specific assays to determine the subtype of VWD.
VWF multimer analysis.
SDS-agarose electrophoresis is used to determine the complement of VWF oligomers in the plasma.16,22 Normal plasma contains dimers up to >40 mers of VWF. Multimers are classified as high, intermediate, or low molecular weight by counting bands 1–5 as low molecular weight, 6–10 as intermediate molecular weight, and >10 as high-molecular-weight (HMW) forms.23 HMW multimers are decreased or missing in types 2A and 2B VWD, and intermediate MW may also be lost in type 2A. Abnormalities in satellite (“triplet”) band patterns can give clues about pathogenesis and help to subclassify type 2A VWD using a tertiary classification10 based on multimeric profile and terminology from an earlier classification,24 in which current type 2A was represented by several subtypes including IIA, IIC, IID and IIE, e.g., type 2A(IIE).22
Ristocetin induced platelet aggregation.
Ability of VWF to aggregate platelets at 2–3 concentrations of ristocetin. Aggregation at a low concentration (∼0.5 mg/mL) is abnormal and may indicate type 2B or its phenocopy, platelet-type pseudo VWD (PT-VWD) resulting from mutations in GPIBA, in which enhanced VWF-platelet binding is present.17,25
Binding of FVIII by VWF (VWF:FVIIIB).
This test that determines the ability of VWF to bind FVIII26,27 is useful in the identification and differential diagnosis of type 2N VWD.28 There are no standard units for the output of this test.
Collagen binding assay (VWF:CB).
This test determines the ability of VWF to bind to collagen (a subendothelial matrix component). It is used in some centers to help identify functional VWF discordance (i.e., to help distinguish types 1 and 2 VWD). Collagen binding is dependent on the presence of HMW multimers; reduced collagen binding reflects the loss of HMW multimer forms29 or can reflect a specific collagen-binding deficiency. The normal range is approximately 50–200 IU/dL.
MOLECULAR GENETIC TESTING
Molecular genetic pathogenesis
The VWF gene spans 178 kb of genomic DNA. Its 52 exons encode an 8.8 kb mRNA and a 2813 amino acid (AA) protein.30–32
Currently, more than 160 normal variants are known in the exons and closely flanking intronic sequences; AA substitutions are reported at 30 residues (Table 2). As non-white ethnic groups are being examined, many additional normal variants are being identified. In one study, an average of 17 heterozygous sequence variants were identified in each individual screened for VWF mutations.33 This high degree of polymorphism in the VWF gene, along with the large size of the gene and the presence of a partial pseudogene, VWFP, replicating exons 23–34 can make full gene sequencing and data interpretation challenging.31 For type 1 VWD, approximately 10–15% of index cases examined in three large muticenter studies had more than one candidate mutation identified; in approximately half of the cases, the variants were in cis (on the same allele), whereas the remainder was in trans.34–36 Such observations underscore the difficulty of identifying pathologic versus normal allelic variants. Several single-nucleotide variants in the VWF promoter region have been identified,34–36 but their pathogenicity has not yet been confirmed.
Normal allelic variants useful for linkage analysis are summarized in Table 2 and include short tandem repeats in the promoter and intron 40 (rs41402545 and rs36115023),37 plus a number of common single-nucleotide variants.
Normal gene product
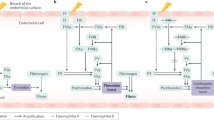
The 2813 AA VWF protein comprises a 22-AA signal peptide, a 741-AA propeptide, and a 2050-AA mature protein.32 Its domain structure from the amino terminus is S-D1-D2-D'-D3-A1-A2-A3-D4-B1-B2-B3-C1-C2-CK (Fig. 1). During synthesis, tail-to-tail disulphide-linked dimers are formed through the CK domains, followed by head-to-head VWF oligomers. Two disulphide isomerase-like sites in the propeptide (CGLC sequences at residues 159–162 and 521–524) facilitate this process.38 VWF has two sites of synthesis; endothelial cells and megakaryocytes, the precursors of platelets. It is stored in endothelial Weible-Palade bodies and platelet alpha granules, being released from alpha granules on platelet activation. When secreted, VWF consists of multimers up to 40 subunits (dimers) in length. The propeptide is cleaved by furin between AAs 763 and 764 during multimer production, and the propeptide (VWFpp) is secreted into the plasma along with VWF. The ratio between VWFpp and mature VWF (VWFpp/VWF:Ag) can be used to estimate relative half-life of mature VWF with elevated ratios indicating enhanced clearance.39,40

Location of VWF mutations by VWD type. Thick horizontal lines indicate the approximate position of exons where mutations are most prevalent, and thinner lines indicate exons with mutations of lower frequency. Mutations that result in type 2 VWD affect VWF function and cluster in domains primarily disrupted by missense mutations.
To render HMW VWF less thrombogenic, it is cleaved by ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type 1 motif)41 between AAs p.Tyr1605-Met1606 after secretion.42 This multimer proteolysis produces the characteristic “triplet” pattern of satellite bands flanking each main multimer band observed on multimer analysis gels. Abnormalities resulting from enhanced or reduced proteolysis can give clues as to VWD subtype.22
Abnormal gene product
Abnormalities in VWF are dependent on the type of mutation; the molecular consequences of both the protein and nucleotide abnormality result in different VWD types.
Type 1
VWF mutations have been identified in up to approximately 65% of patients diagnosed with type 1 VWD in multicenter studies.34–36 Missense mutations predominate but may affect VWF through different mechanisms. A common heterozygous in-frame large deletion of exons 4–5 has recently been reported in a cohort of UK patients with type 1 VWD,43 and this along with similar large deletions may also contribute to the mutation spectrum in this disease type. As molecular genetic testing has only been undertaken in type 1 VWD relatively recently, pathogenic mechanisms have not yet been ascertained in many cases.
Missense mutations mostly in the D3 domain39,44 reduce the residence time of VWF in plasma by up to 15-fold. p.Arg1205His, the so-called “Vicenza” variant,45–47 is the best characterized and most common of these mutations.34–36 Such mutations have been referred to as type 1 clearance (1C),40 although this is not a VWD category that has been recognized in the ISTH SSC on VWF classification.10 Intracellular retention seems to be a common mechanism for type 1 VWD pathogenicity and has recently been demonstrated for missense mutations in a number of VWF domains.48 Haploinsufficiency resulting from a heterozygous null allele results in reduced VWF expression in a small proportion of cases.34–36
Type 2A
This subtype is the most common among type 2 VWD variants and encompasses missense mutations in several locations, which act through a variety of mechanisms to result in a loss of HMW and sometimes intermediate molecular weight multimers: (1) impaired dimer assembly (CK domain); (2) impaired multimer assembly (D3 and D2 domains)49; (3) enhanced susceptibility to ADAMTS13 (A2 and A1 domains)50; (4) intracellular retention (D3, A1, and A2 domains).49 More than one mechanism can contribute to the pathogenesis of a number of mutations. All result in VWF with fewer GpIbα binding sites and less effective platelet clot formation. The mutations are classified as a group, as clinically affected individuals are treated similarly.10
Type 2B
Missense mutations enhance the ability of VWF to bind platelet glycoprotein GpIbα, such that binding occurs spontaneously without the requirement for VWF to bind to collagen after subendothelial damage. The resulting platelet-VWF complex is removed from circulation and can result in thrombocytopenia. The largest multimers bind platelets preferentially, so are lost to a greater extent. VWF binding to platelets can also enhance its susceptibility to ADAMTS13 cleavage, which also contributes to the loss of HMW multimers. Although all these features may be present to varying degrees in the majority of patients, not all cases demonstrate the “classic” 2B VWD presentation. For example, mutations affecting p.Pro1266Leu may only demonstrate enhanced GpIbα binding (ristocetin induced platelet aggregation) but no thrombocytopenia or HMW multimer loss,18 and in some cases, all parameters other than ristocetin induced platelet aggregation may be normal.51,52
Type 2M
VWF is poor at binding GpIbα, often because of mutations in the A1 domain that alter protein conformation and reduce binding affinity (Fig. 1), but without the loss of HMW multimers seen in type 2A.53 Mutations in the A3 domain that affect binding to collagen in the subendothelium54,55 are also classified as 2M VWD.10
Type 2N
Affinity of VWF for FVIII is reduced due to alteration of key AAs in the FVIII binding site or to conformational changes having an indirect effect on VWF-FVIII binding. The majority of patients have a normal multimeric profile, but in some cases, supranormal multimers are present, whereas others may demonstrate a loss of HMW multimers and can have a 2A(IIE) appearance.56
Type 3
Both alleles are affected by mutations (null and missense) that result in lack of VWF secretion from the cell. The majority of patients with type 3 VWD have two null alleles and produce no significant quantity of VWF. Approximately 20% of alleles carry missense mutations predominantly located in the D1–D2 (exons 3–11) domains and D4-CK (exons 37–52) domains (Fig. 1). These may impair dimerization or multimerization of VWF, resulting in intracellular retention and lack of VWF secretion into plasma.57 The remaining 80% are null alleles located throughout VWF. Null alleles can result from many different types of mutations. These mutations do not result in a functional protein product as the mutation results in either the complete absence/instability of mRNA or protein or the expression of a nonfunctional gene product (e.g., a protein that cannot be secreted). Large deletions predominantly resulting in frameshift mutations affecting one or more exons also contribute to type 3 VWD mutation spectrum.58
Currently, VWF is the only gene known to be associated with VWD. However, the ISTH SSC on VWF classification scheme10 does not restrict VWD to being caused by mutations in the VWF gene. Evaluation of the entire protein coding and flanking intronic regions of the VWF gene fails to identify a VWF mutation in some cases of “apparent” VWD. Failure to identify a causative mutation in the VWF gene does not exclude “VWD.” PT-VWD results from mutations in the GPIBA gene but presents phenotypically as type 2B VWD.
CLINICAL TESTING
The domain structure and exons encoding each VWF domain are shown in Figure 1. Sequence analysis of the entire coding region and flanking exon/intron junctions of VWF is available, although it is technically challenging due to the gene structure. Heterozygous large deletion mutations are not detected by polymerase chain reaction and DNA sequencing but require either a deletion-specific gap polymerase chain reaction43 or a dosage analysis technique that can detect any exon deletion/duplication such as multiplex ligation-dependent probe amplification (MLPA).59,60 Linkage analysis can be useful in type 3 VWD to facilitate prenatal diagnosis if time is insufficient for full mutation analysis or if two mutations cannot be identified in an affected individual.61
In those individuals with types 2A and 2B VWD in whom specific VWD hemostasis factor assays can provide a clear diagnosis, molecular genetic testing may not be warranted.62 Molecular genetic testing for VWD may be indicated in the following circumstances.
(1) To establish the VWD subtype in those individuals in whom specific VWD hemostasis factor assays suggest VWD but in whom genetic analysis may provide a more definitive diagnosis; (2) to distinguish between type 2N VWD, mild hemophilia A (males), or (symptomatic) carriers of hemophilia A (females) with reduced FVIII:C levels, where phenotypic testing remains inconclusive (e.g., VWF:FVIIIB assay is unavailable or unhelpful); and (3) families with type 3 VWD requesting carrier analysis and prenatal diagnosis. A recent guideline on VWD genetic testing has been published by the UK Hemophilia Centre Doctors Organization.61
Type 1 VWD
Point mutations have been identified in approximately 65% of individuals with type 1 VWD and occur throughout the VWF gene.34–36 Fully penetrant, dominantly inherited missense mutations are more often identified when VWF:Ag and VWF:RCo levels are <25 IU/dL. In contrast, incompletely penetrant dominantly inherited missense mutations, such as p.Tyr1584Cys63 and p.Arg924Gln,64,65 are identified in approximately 50% of individuals whose VWF:Ag and VWF:RCo levels are above 25 IU/dL.
The extent to which incompletely penetrant VWF mutations contribute to bleeding phenotype in individuals with VWF levels of approximately 50 IU/dL is not clear, and genetic analysis in such cases may not be easy to interpret.61
Type 2 VWD
Many mutations seen in types 2A and 2M, and all missense mutations in type 2B are located in exon 28; thus, this exon should be examined first when any of these three VWD subtypes is suspected.
Type 2A VWD
This subtype includes missense mutations acting through a variety of mechanisms giving rise to similar phenotypes. The majority of type 2A VWD is inherited in an autosomal dominant (AD) fashion. The major mutation location is exon 28, where mutations predominantly affect the A2 domain. Missense mutations have also been reported very recently in the D3 and A1 domains (exons 22 and 25–28), resulting in the subtype 2A(IIE). This variant, which is reported to make up approximately 30% of all 2A cases, includes many missense mutations affecting cysteine residue.49 Less common missense mutations responsible for dimerization (CK domain, exon 52, 2A[IID])58 and multimerization defects (D2 domain, exons 11–17, 2A[IIC])58,66 can be sought subsequently. In the rare, recessive form of the disorder (2A[IIC]), affected individuals are either homozygous for the same missense mutation (often seen in consanguineous families) or compound heterozygous for a missense mutation and a null allele.66 Presence of a second null mutation may be inferred, unless the full gene is analyzed.58
Type 2B VWD
Missense mutations are located in exon 28 in or close to the A1 domain.18
Type 2M VWD
A small number of missense mutations are reported in exon 28, with case reports in exons 27, 30–31, and 52.54,55,58 These additional locations can be analyzed after exon 28.
Type 2N VWD
The majority (∼80%) of missense mutations are located in exons 18–20, which can be analyzed first with a much lower proportion of mutations in exons 17 and 24–27.28,58 Type 2N has autosomal recessive (AR) inheritance, and affected individuals are either homozygous or compound heterozygous for missense mutations, or compound heterozygous for a missense mutation and a mutation resulting in lack of VWF expression. The latter mutations are found throughout VWF. p.Arg854Gln occurs in 2% of white populations66,67 and frequently contributes to the phenotype. In an individual referred for genetic testing, lack of a missense mutation in the above exons can exclude VWD 2N.
Type 3 VWD
Mutations associated with type 3 VWD are found throughout the entire coding region of VWF (i.e., exons 2–52). Sequence analysis of this region identifies mutations in 80–90% of patients with type 3 VWD. Deletion/duplication (dosage) analysis has recently become available using MLPA,59 but the extent of contribution of large deletions or duplications has not been established within type 3 VWD patient cohorts, and large heterozygous mutations have likely been overlooked. Large deletions currently comprise 9% of mutation reports in patients with type 3 VWD on VWFdb.58 Linkage analysis can be useful in type 3 VWD to facilitate prenatal diagnosis if time is insufficient for full mutation analysis or if two mutations cannot be identified in an affected individual.61
CLINICAL DESCRIPTION
Natural history
VWD is a congenital bleeding disorder; however, symptoms may only become apparent on hemostatic challenge, and bleeding history may become more obvious with increasing age. Thus, it may take some time before an abnormal bleeding history is recognized. Bleeding history is also dependent on disease severity; type 3 VWD is often diagnosed early in life, but in families with no disease history, nonaccidental injury may be suspected, whereas mild type 1 VWD may not be diagnosed until midlife, despite a history of bleeding episodes. Individuals with VWD primarily manifest excessive mucocutaneous bleeding (bruising, epistaxis, menorrhagia, etc.) and do not tend to experience musculoskeletal bleeding unless their FVIII:C level is <10 IU/dL, as can be seen in type 2N or type 3 VWD.
Type 1 VWD
Type 1 VWD accounts for up to 70% of all VWD. It typically manifests as mild mucocutaneous bleeding; however, symptoms can be more severe when VWF levels are <15 IU/dL. Epistaxis and bruising are common symptoms among children. Menorrhagia is the most common finding in women of reproductive age.68,69
Type 2 VWD
Type 2 VWD accounts for approximately 25% of all VWD. The relative frequency of the subtypes is 2A > 2N > 2M > 2B in European populations.
Individuals with type 2A VWD usually present with mild to moderate mucocutaneous bleeding.
Patients with type 2B VWD typically present with mild to moderate mucocutaneous bleeding, although in some cases it can be severe.70 In many patients with type 2B VWD, the development or worsening of thrombocytopenia may occur during stressful situations, such as severe infection, during surgery or pregnancy, or if treated with desmopressin.18
In type 2M VWD, individuals typically present with mild to moderate mucocutaneous bleeding symptoms, but bleeding episodes can be severe, particularly in the presence of very low or absent VWF:RCo.
Symptoms of type 2N VWD are essentially the same as those seen in mild hemophilia A and include excessive bleeding at the time of surgery or procedures as both disorders result from reduced FVIII:C.
Type 3 VWD
Type 3 VWD accounts for <5% of VWD. It manifests with severe bleeding including both excessive mucocutaneous bleeding and musculoskeletal bleeding. The latter may be more pronounced where FVIII:C levels are lowest.
Penetrance
In AD type 1 VWD, mutations resulting in plasma VWF level <25 IU/dL are often fully penetrant, whereas those resulting in higher VWF levels are often incompletely penetrant. Mutations causal for other AD VWD types; 2A, 2B, and 2M are often fully penetrant.
DIFFERENTIAL DIAGNOSIS
Both type 2N VWD and mild hemophilia A (caused by mutations in F8) result in reduced levels of FVIII:C (∼5 to 40 IU/dL) with normal or borderline low levels of VWF. Although the VWF:FVIIIB test, which determines the ability of VWF to bind FVIII, is a useful way to distinguish between the two disorders,71 its availability may be limited and the test may sometimes yield equivocal results.72
In families with reduced FVIII:C, an X-linked pattern of inheritance can help identify those with mild hemophilia A. When family history is uninformative, it may be preferable to perform sequence analysis of F8 before VWF, even in symptomatic females who are simplex cases (i.e., a single occurrence in a family), because a F8 mutation and skewed X-chromosome inactivation (Lyonization) are often responsible for symptoms. In these cases, F8 intrachromosomal inversions may be sought and DNA sequence analysis or mutation scanning of F8 exons 1–26 undertaken.73,74 In females, dosage analysis using MLPA can also be used to identify heterozygous partial or complete gene deletions or duplications.75,76 F8 mutations may be detected in >50% of cases referred for “possible 2N VWD or hemophilia A.”72 When F8 mutations are absent, VWF can be analyzed.
PT-VWD (also called pseudo VWD) mimics type 2B VWD but is caused by mutations in GPIBA. The disorders can be distinguished by mixing patient or control plasma and platelets to determine which component is defective using platelet aggregation or flow cytometry, but these assays are technically challenging.25,52,77,78 When mutations are absent from exon 28 of VWF, mutations in exon 2 of GPIBA may be identified. To date, missense mutations reported to affect GpIbα AAs p.Gly249, and p.Met255 plus a 27bp in-frame deletion p.Pro449_Ser457del (c.1345_ 1371del27)79,80 have been reported (following Human Genome Variation Society nomenclature81; reference sequences NP_000164.4 and NM_000173.4, legacy numbering G233, M239, P433–441 deletion).
PT-VWD is probably underdiagnosed.79 Misdiagnosis of PT-VWD may result in ineffective treatment of patients. VWF concentrate is needed to correct reduced VWF level, but platelet transfusion may also be required if there is significant thrombocytopenia.25 The half-life of replaced VWF is reduced in PT-VWD due to binding to abnormal GpIbα, so VWF concentrate has to be administered more frequently than in VWD. Molecular genetic testing of GPIBA may identify missense or in-frame mutations80,82 in approximately 10% of persons diagnosed with 2B VWD.
Acquired von Willebrand syndrome
This mild to moderate bleeding disorder can occur due to a variety of conditions83–85 but is not caused by VWF mutation. It is most often seen in persons older than 40 years with no bleeding history. Acquired von Willebrand syndrome has diverse pathology and may result from (1) lymphoproliferative or plasma cell proliferative disorders, paraproteinemias (monoclonal gammopathy of unknown significance), multiple myeloma, and Waldenstrom macroglobulinemia. Antibodies against VWF have been detected in some of these cases: (2) autoimmune disorders including systemic lupus erythrematosus, scleroderma, and antiphospholipid antibody syndrome; (3) shear-induced VWF conformational changes leading to increased VWF proteolysis (such as aortic valvular stenosis and ventricular septal defect); (4) markedly increased blood platelet count (including essential thrombocythemia or other myeloproliferative disorders); (5) removal of VWF from circulation by aberrant binding to tumor cells (e.g., Wilms tumor or certain lymphoproliferative disorders); (6) decreased VWF synthesis (e.g., resulting from hypothyroidism); and (7) use of certain drugs (including valproic acid, ciprofloxacin, griseofulvin, and hydroxyethyl starch).
MANAGEMENT
Evaluations after initial diagnosis
To establish the extent of disease in an individual diagnosed with VWD, the following evaluations are recommended: (1) a personal and family history of bleeding to help predict severity and tailor treatment. Use of a standardized bleeding assessment tool can be helpful.13,14,86 (2) A joint and muscle evaluation for those with type 3 VWD (musculoskeletal bleeding is rare in types 1 and 2 VWD). (3) Screening for hepatitis B and hepatitis C as well as HIV if the diagnosis is type 3 VWD or if the individual received blood products or plasma-derived clotting factor concentrates before 1985. (4) Baseline serum concentration of iron and ferritin (to assess iron stores), as many individuals with VWD are iron deficient, particularly women with menorrhagia. (5) A gynecological evaluation for women with menorrhagia.87
Treatment of manifestations
Individuals with VWD benefit from referral to a comprehensive bleeding disorders program for education, treatment, and genetic counseling.
The two main treatments are desmopressin (1-deamino-8-d-arginine vasopressin [DDAVP]) and clotting factor concentrates containing both VWF and FVIII (VWF/FVIII concentrate). Individuals with VWD should receive prompt treatment for severe bleeding episodes.
Desmopressin
Most individuals with type 1 VWD and some with type 2 VWD respond to intravenous or subcutaneous treatment with desmopressin,88,89 which promotes release of stored VWF and raises levels 3- to 10-fold. Intranasal preparations are also available. Following VWD diagnosis, a desmopressin challenge is advisable to assess VWF response.17 Desmopressin is the treatment of choice for acute bleeding episodes or to prevent bleeding during surgery. In persons who are intolerant to desmopressin or have a poor VWF response, clotting factor concentrate is required. Desmopressin is contraindicated in individuals with arteriovascular disease and in those older than 70 years for whom VWF/FVIII concentrate is required. Because desmopressin can cause hyponatremia (imbalance of water to salt, which can lead to seizures and coma), fluid intake should be restricted for 24 hours after its administration to minimize this risk.
Intravenous infusion of VWF/FVIII clotting factor concentrates
In those who are nonresponsive to desmopressin (i.e., where the VWF deficiency is not sufficiently corrected) and for those in whom desmopressin is contraindicated (below), bleeding episodes can be prevented or controlled with intravenous infusion of virally inactivated plasma-derived clotting factor concentrates containing both VWF and FVIII.90 Such concentrates are prepared from pooled blood donations from many donors. Virus inactivation procedures eliminate potential pathogens.
Indirect treatments
In addition to treatments that directly increase VWF levels, individuals with VWD often benefit from indirect hemostatic treatments. Fibrinolytic inhibitors (i.e., tranexamic acid) can be helpful for treatment or prevention of bleeding episodes.91 Hormonal treatments (i.e., the combined oral contraceptive pill) can be particularly effective for the treatment of menorrhagia.92 Nonmedical treatments including the levonorgestrel-releasing intrauterine system93 or endometrial ablation94 may be useful in selected patients with VWD. A consensus document on the management of women with VWD was recently published.95
Obstetric issues
VWF levels increase throughout pregnancy with the peak occurring in the third trimester.96 Despite this, pregnant women with VWD are at increased risk for bleeding complications, and care should be provided in centers with experience in perinatal management of bleeding disorders.12,97
Although deliveries should occur based on obstetric indications, instrumentation should be minimized.87 Delayed, secondary postpartum bleeding may be a problem as VWF level rapidly returns to prepregnancy level after delivery.
Pediatric issues
Special considerations should be taken for the care of infants and children with VWD.
Infant males should only be circumcised after consultation with a pediatric hemostasis specialist. Desmopressin should be used with caution, particularly in those younger than 2 years, because of the potential difficulty in restricting fluids in this age group.
Additionally, VWF levels are higher in the neonatal period, and so phenotypic testing for milder forms of VWD should be delayed until later in childhood.
Prevention of primary manifestations
Individuals with type 3 VWD may be given prophylactic infusions of VWF/FVIII concentrates to prevent musculoskeletal bleeding and subsequent joint damage.98
Prevention of secondary complications
Desmopressin should be used with caution, particularly in those younger than 2 years. Individuals with VWD should be vaccinated for hepatitis A and hepatitis B.17 Prevention of chronic joint disease is a concern for individuals with type 3 VWD; however, controversy exists about the specific schedule and dosing of prophylactic regimens.98 This is the subject of an ongoing international trial, with prophylactic treatment being studied for joint bleeding, nosebleeds, and menorrhagia.
Surveillance
Individuals with milder forms of VWD can benefit from being followed by treatment centers with experience in the management of bleeding disorders. Individuals with type 3 VWD should be followed up in experienced centers and should have periodic evaluations by a physiotherapist to monitor joint mobility.
Agents/circumstances to avoid
Activities with a high risk of trauma, particularly head injury should be avoided. Medications with effects on platelet function (aspirin [acetylsalicylic acid], clopidogrel, or nonsteroidal antiinflammatory drugs) should be avoided as they can worsen bleeding symptoms. Infant males should only be circumcised after consultation with a pediatric hemostasis specialist.
THERAPIES UNDER INVESTIGATION
Recombinant VWF is in clinical trials99 and is expected to be available for patient use shortly.
GENETIC COUNSELING
Mode of inheritance
Most cases of VWD type 1, along with types 2A, 2B, and 2M are inherited in an AD manner, whereas types 2N, 3, and rarer occurrences of types 1 and 2A are inherited in an AR manner.
Risk to family members—AD inheritance
Parents of a proband
Most individuals diagnosed with one of the AD types of VWD have an affected parent. However, a proband with AD VWD may have the disorder as the result of a new gene mutation. The proportion of cases caused by de novo mutations has not been well studied, but recent multicenter studies on type 1 VWD identified de novo mutations in at least 2–4% of probands, which were absent from parents.34,100 Where the mutation causing AD VWD found in the proband cannot be detected in the DNA of either parent, two possible explanations are germline mosaicism in a parent and a de novo mutation in the proband. Neither possibility has been sufficiently investigated to comment on relative likelihood of occurrence. Evaluation of parents of a proband with AD VWD may determine that one is affected but has escaped previous diagnosis because of failure by health care professionals to recognize the symptoms and/or a milder phenotypic presentation. Therefore, an apparently negative family history cannot be confirmed until appropriate evaluations have been performed.
Siblings of a proband
The risk to the siblings (sibs) of the proband depends on the genetic status of the proband's parents. Where the parent of the proband is affected, the risk to the sibs is 50%. The sibs of a proband with clinically unaffected parents are still at increased risk for VWD because of the possibility of reduced penetrance in a parent. If the disease-causing mutation found in the proband cannot be detected in the DNA of either parent, then the risk to sibs is low, but greater than that of the general population, because of the possibility of germline mosaicism.
Offspring of a proband
Each child of an individual with AD VWD has a 50% chance of inheriting the mutation.
Other family members of a proband
The risk to other family members depends on the status of the proband's parents. If a parent is found to be affected, his or her family members may be at risk.
Risk to family members—AR inheritance
Parents of a proband
The parents of an individual with AR VWD are obligate heterozygotes (i.e., carriers of one mutant allele). Heterozygotes (carriers) are generally asymptomatic. For type 3 VWD, approximately 15% show some mild bleeding symptoms, whereas more than 50% have VWF levels <50 IU/dL.17 Reduced FVIII level correlates with increased bleeding tendency and individuals with low levels may be more at risk of bleeding associated with surgery.101
Sibs of a proband
At conception, each sib of an individual with AR VWD has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once an at-risk sib is known to be unaffected, the risk of his/her being a carrier is 2/3. Heterozygotes (carriers) are generally asymptomatic.
Offspring of a proband
The offspring of an individual with AR VWD is obligate heterozygote (carrier) for a disease-causing mutation in the VWF gene.
Other family members of a proband
Each sib of the proband's parents is at 50% risk of being a carrier.
Carrier detection
Carrier testing for at-risk family members is possible once the disease-causing mutations have been identified in the family.
Prenatal testing
Prenatal diagnosis for pregnancies at increased risk (generally only for type 3 VWD) is possible by analysis of DNA extracted from fetal cells obtained by chorionic villus sampling at approximately 11–13 weeks gestation or amniocentesis usually performed at approximately 15–18 weeks gestation. The disease-causing allele(s) of an affected family member must be identified before prenatal testing can be performed. Preimplantation genetic diagnosis may become available for families in which the disease-causing mutation(s) have been identified.102,103
GENOTYPE-PHENOTYPE CORRELATIONS
Response to desmopressin
Correlation of mutation with the response to treatment has been investigated in types 1 and 2 VWD. Most of 77 EU patients with type 1 VWD demonstrated clinically useful rises in VWF and FVIII levels after desmopressin infusion, those who did not largely had mutations in the D3 and A1–A3 domains.88 In type 2 VWD, many patients have a clinically useful response to desmopressin, although it may be short lived in some cases. In type 2B cases, however, thrombocytopenia may be exacerbated.18,104 However, individuals with 2B mutations p.Pro1266Leu and p.Arg1308Leu do not experience thrombocytopenia and can be treated with desmopressin.18 Utility of desmopressin for management of delivery in types 1 and 2 VWD has recently been correlated with causative mutations, with prevention of significant bleeding by 2–3 administrations in most cases.96
Inhibitory antibodies
A small proportion of patients with type 3 VWD develop antibodies in response to replacement therapy (7.5–9.5%).105,106 Systematic surveys of mutation correlation with inhibitors have not been conducted, but large gene deletions and nonsense mutations have been reported in this patient group. Anaphylaxis after replacement therapy may also be experienced by the same individuals.105
ABO blood group
Blood group contributes approximately 25% of the variance in plasma VWF level; ABO glycosylation of VWF influences its rate of clearance.107 Individuals with non-O blood groups have higher VWF levels than those with O blood group and those with group AB have the highest levels. Blood group O is significantly more common in type 1 VWD than in the general population or in type 2 VWD108 and is particularly common in cases with no mutation identified.35 ABO blood group seems to be an important contributor to penetrance and reduced VWF level in type 1 VWD34,35 as has been observed with the common mutation p.Tyr1584Cys.63,109
Nomenclature
Current and previously used nomenclature for VWD and VWF is detailed in Table 4.
CONCLUDING COMMENTS
Significant progress has been made in our understanding of VWD since its original description in 1926 by Dr. Erik von Willebrand. The identification of VWF, and subsequently, the identification, cloning, and characterization of the VWF gene have led to the availability of genetic testing, which when rationally applied can aid in the management of patients.
REFERENCES
von Willebrand EA . Hereditar pseudohemofili. Finska Lakarsallskapets Handl 1926; 67: 7–112.
Lynch DC, Zimmerman TS, Collins CJ, et al. Molecular cloning of cDNA for human von Willebrand factor: authentication by a new method. Cell 1985; 41: 49–56.
Sadler JE, Shelton-Inloes BB, Sorace JM, et al. Cloning and characterization of two cDNAs coding for human von Willebrand factor. Proc Natl Acad Sci USA 1985; 82: 6394–6398.
Ginsburg D, Handin RI, Bonthron DT, et al. Human von Willebrand factor (vWF): isolation of complementary DNA (cDNA) clones and chromosomal localization. Science 1985; 228: 1401–1406.
Verweij CL, Diergaarde PJ, Hart M, Pannekoek H . Full-length von Willebrand factor (vWF) cDNA encodes a highly repetitive protein considerably larger than the mature vWF subunit. EMBO J 1986; 5: 1839–1847.
Bowman M, Hopman WM, Rapson D, et al. A prospective evaluation of the prevalence of symptomatic von Willebrand disease (VWD) in a pediatric primary care population. Pediatr Blood Cancer 2010; 55: 171–173.
Rodeghiero F, Castaman G, Dini E . Epidemiological investigation of the prevalence of von Willebrand's disease. Blood 1987; 69: 454–459.
Werner EJ, Broxson EH, Tucker EL, et al. Prevalence of von Willebrand disease in children: a multiethnic study. J Pediatr 1993; 123: 893–898.
Bloom AL . von Willebrand factor: clinical features of inherited and acquired disorders. Mayo Clin Proc 1991; 66: 743–751.
Sadler JE, Budde U, Eikenboom JC, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand factor. J Thromb Haemost 2006; 4: 2103–2114.
Rodeghiero F . Management of menorrhagia in women with inherited bleeding disorders: general principles and use of desmopressin. Haemophilia 2008; 14: suppl 1 21–30.
James AH, Jamison MG . Bleeding events and other complications during pregnancy and childbirth in women with von Willebrand disease. J Thromb Haemost 2007; 5: 1165–1169.
Tosetto A, Rodeghiero F, Castaman G, et al. A quantitative analysis of bleeding symptoms in type 1 von Willebrand disease: results from a multicenter European study (MCMDM-1 VWD). J Thromb Haemost 2006; 4: 766–773.
Bowman M, Mundell G, Grabell J, et al. Generation and validation of the Condensed MCMDM-1VWD Bleeding Questionnaire for von Willebrand disease. J Thromb Haemost 2008; 6: 2062–2066.
Bowman M, Riddel J, Rand ML, et al. Evaluation of the diagnostic utility for von Willebrand disease of a pediatric bleeding questionnaire. J Thromb Haemost 2009; 7: 1418–1421.
Budde U . Diagnosis of von Willebrand disease subtypes: implications for treatment. Haemophilia 2008; 14: suppl 5 27–38.
Nichols WL, Hultin MB, James AH, et al. von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). Haemophilia 2008; 14: 171–232.
Federici AB, Mannucci PM, Castaman G, et al. Clinical and molecular predictors of thrombocytopenia and risk of bleeding in patients with von Willebrand disease type 2B: a cohort study of 67 patients. Blood 2009; 113: 526–534.
Podda GM, Bucciarelli P, Lussana F, Lecchi A, Cattaneo M . Usefulness of PFA-100 testing in the diagnostic screening of patients with suspected abnormalities of hemostasis: comparison with the bleeding time. J Thromb Haemost 2007; 5: 2393–2398.
Favaloro EJ . Laboratory monitoring of therapy in von Willebrand disease: efficacy of the PFA-100 and von Willebrand factor:collagen-binding activity as coupled strategies. Semin Thromb Hemost 2006; 32: 566–576.
Castaman G, Tosetto A, Goodeve A, et al. The impact of bleeding history, von Willebrand factor and PFA-100 on the diagnosis of type 1 von Willebrand disease: results from the European study MCMDM-1VWD. Br J Haematol 2010; 151: 245–251.
Budde U, Pieconka A, Will K, Schneppenheim R . Laboratory testing for von Willebrand disease: contribution of multimer analysis to diagnosis and classification. Semin Thromb Hemost 2006; 32: 514–521.
ISTH-VWF-SSC. Minutes of the von Willebrand factor subcommittee, 2004; Available at: www.isth.org/default/index.cfm/publication/ssc-minutes. Accessed January 10, 2011.
Ruggeri ZM, Zimmerman TS . von Willebrand factor and von Willebrand disease. Blood 1987; 70: 895–904.
Franchini M, Montagnana M, Lippi G . Clinical, laboratory and therapeutic aspects of platelet-type von Willebrand disease. Int J Lab Hematol 2008; 30: 91–94.
Caron C, Mazurier C, Goudemand J . Large experience with a factor VIII binding assay of plasma von Willebrand factor using commercial reagents. Br J Haematol 2002; 117: 716–718.
Zhukov O, Popov J, Ramos R, et al. Measurement of von Willebrand factor-FVIII binding activity in patients with suspected von Willebrand disease type 2N: application of an ELISA-based assay in a reference laboratory. Haemophilia 2009; 15: 788–796.
Mazurier C, Hilbert L . Type 2N von Willebrand disease. Curr Hematol Rep 2005; 4: 350–358.
Favaloro EJ . Von Willebrand factor collagen-binding (activity) assay in the diagnosis of von Willebrand disease: a 15-year journey. Semin Thromb Hemost 2002; 28: 191–202.
Titani K, Kumar S, Takio K, et al. Amino acid sequence of human von Willebrand factor. Biochemistry 1986; 25: 3171–3184.
Mancuso DJ, Tuley EA, Westfield LA, et al. Structure of the gene for human von Willebrand factor. J Biol Chem 1989; 264: 19514–19527.
Sadler JE . Biochemistry and genetics of von Willebrand factor. Annu Rev Biochem 1998; 67: 395–424.
Soteh MH, Peake IR, Marsden L, et al. Mutational analysis of the von Willebrand factor gene in type 1 von Willebrand disease using conformation sensitive gel electrophoresis: a comparison of fluorescent and manual techniques. Haematologica 2007; 92: 550–553.
James PD, Notley C, Hegadorn C, et al. The mutational spectrum of type 1 von Willebrand disease: results from a Canadian cohort study. Blood 2007; 109: 145–154.
Goodeve A, Eikenboom J, Castaman G, et al. Phenotype and genotype of a cohort of families historically diagnosed with type 1 von Willebrand disease in the European study, Molecular and Clinical Markers for the Diagnosis and Management of Type 1 von Willebrand disease (MCMDM-1VWD). Blood 2007; 109: 112–121.
Cumming A, Grundy P, Keeney S, et al. An investigation of the von Willebrand factor genotype in UK patients diagnosed to have type 1 von Willebrand disease. Thromb Haemost 2006; 96: 630–641.
Vidal F, Julia A, Altisent C, Puig L, Gallardo D . Von Willebrand gene tracking by single-tube automated fluorescent analysis of four short tandem repeat polymorphisms. Thromb Haemost 2005; 93: 976–981.
Mayadas TN, Wagner DD . Vicinal cysteines in the prosequence play a role in von Willebrand factor multimer assembly. Proc Natl Acad Sci USA 1992; 89: 3531–3535.
Haberichter SL, Castaman G, Budde U, et al. Identification of type 1 von Willebrand disease patients with reduced von Willebrand factor survival by assay of the VWF propeptide in the European study: molecular and clinical markers for the diagnosis and management of type 1 VWD (MCMDM-1VWD). Blood 2008; 111: 4979–4985.
Haberichter SL, Balistreri M, Christopherson P, et al. Assay of the von Willebrand factor (VWF) propeptide to identify patients with type 1 von Willebrand disease with decreased VWF survival. Blood 2006; 108: 3344–3351.
Zheng X, Chung D, Takayama TK, et al. Structure of von Willebrand factor-cleaving protease (ADAMTS13), a metalloprotease involved in thrombotic thrombocytopenic purpura. J Biol Chem 2001; 276: 41059–41063.
Feys HB, Anderson PJ, Vanhoorelbeke K, Majerus EM, Sadler JE . Multi-step binding of ADAMTS-13 to von Willebrand factor. J Thromb Haemost 2009; 7: 2088–2095.
Sutherland MS, Cumming AM, Bowman M, et al. A novel deletion mutation is recurrent in von Willebrand disease types 1 and 3. Blood 2009; 114: 1091–1098.
Millar CM, Riddell AF, Brown SA, et al. Survival of von Willebrand factor released following DDAVP in a type 1 von Willebrand disease cohort: influence of glycosylation, proteolysis and gene mutations. Thromb Haemost 2008; 99: 916–924.
Mannucci PM, Lombardi R, Castaman G, et al. von Willebrand disease “Vicenza” with larger-than-normal (supranormal) von Willebrand factor multimers. Blood 1988; 71: 65–70.
Goodeve AC . Vicenza deciphered: modelling the von Willebrand disease enigma Commentary on Accelerated clearance alone explains ultralarge multimers in VWD Vicenza. J Thromb Haemost 2010; 8: 1271–1272.
Schneppenheim R, Federici AB, Budde U, et al. Von Willebrand Disease type 2M “Vicenza” in Italian and German patients: identification of the first candidate mutation (G3864A; R1205H) in 8 families. Thromb Haemost 2000; 83: 136–140.
Eikenboom J, Hilbert L, Ribba AS, et al. Expression of 14 von Willebrand factor mutations identified in patients with type 1 von Willebrand disease from the MCMDM-1VWD study. J Thromb Haemost 2009; 7: 1304–1312.
Schneppenheim R, Michiels JJ, Obser T, et al. A cluster of mutations in the D3 domain of von Willebrand factor correlates with a distinct subgroup of von Willebrand disease: type 2A/IIE. Blood 2010; 115: 4894–4901.
Hassenpflug WA, Budde U, Obser T, et al. Impact of mutations in the von Willebrand factor A2 domain on ADAMTS13-dependent proteolysis. Blood 2006; 107: 2339–2345.
Casonato A, Sartorello F, Pontara E, et al. A novel von Willebrand factor mutation (I1372S) associated with type 2B-like von Willebrand disease: an elusive phenotype and a difficult diagnosis. Thromb Haemost 2007; 98: 1182–1187.
Favaloro EJ . von Willebrand disease, type 2B: a diagnosis more elusive than previously thought. Thromb Haemost 2008; 99: 630–631; author reply 32–33.
James PD, Notley C, Hegadorn C, et al. Challenges in defining type 2M von Willebrand disease: results from a Canadian cohort study. J Thromb Haemost 2007; 5: 1914–1922.
Riddell AF, Gomez K, Millar CM, et al. Characterization of W1745C and S1783A: 2 novel mutations causing defective collagen binding in the A3 domain of von Willebrand factor. Blood 2009; 114: 3489–3496.
Ribba AS, Loisel I, Lavergne JM, et al. Ser968Thr mutation within the A3 domain of von Willebrand factor (VWF) in two related patients leads to a defective binding of VWF to collagen. Thromb Haemost 2001; 86: 848–854.
Goodeve AC . The genetic basis of von Willebrand disease. Blood Rev 2010; 24: 123–134.
Allen S, Goodeve AC, Peake IR, Daly ME . Endoplasmic reticulum retention and prolonged association of a von Willebrand's disease-causing von Willebrand factor variant with ERp57 and calnexin. Biochem Biophys Res Commun 2001; 280: 448–453.
VWFdb. International Society on Thrombosis and Haemostasis Scientific and Standardization Committee VWF Information Homepage. Available at: www.vwf.group.shef.ac.uk. Accessed January 10, 2011.
Acquila M, Bottini F, DI Duca M, Vijzelaar R, Molinari AC, Bicocchi MP . Multiplex ligation-dependent probe amplification to detect a large deletion within the von Willebrand gene. Haemophilia 2009; 15: 1346–1348.
Hampshire DJ, Bloomer LD, Al-Baba S, et al. Validation of multiplex ligation-dependent probe amplification (MLPA) to detect large-scale deletions within the von Willebrand factor (VWF) gene. Platelets 2009; 20: suppl 1 13–14.
Keeney S, Bowen D, Cumming A, et al. The molecular analysis of von Willebrand disease: a guideline from the UK Haemophilia Centre Doctors' Organisation Haemophilia Genetics Laboratory Network. Haemophilia 2008; 14: 1099–1111.
Favaloro EJ . Genetic testing for von Willebrand disease: the case against. J Thromb Haemost 2009; 8: 6–12.
O'Brien LA, James PD, Othman M, et al. Founder von Willebrand factor haplotype associated with type 1 von Willebrand disease. Blood 2003; 102: 549–557.
Berber E, James PD, Hough C, Lillicrap D . An assessment of the pathogenic significance of the R924Q von Willebrand factor substitution. J Thromb Haemost 2009; 7: 1672–1679.
Hickson N, Hampshire D, Winship P, et al. von Willebrand factor variant p.Arg924Gln marks an allele associated with reduced von Willebrand factor and factor VIII levels. J Thromb Haemost 2010; 8: 1986–1993.
Meyer D, Fressinaud E, Gaucher C, et al. Gene defects in 150 unrelated French cases with type 2 von Willebrand disease: from the patient to the gene. INSERM Network on Molecular Abnormalities in von Willebrand disease. Thromb Haemost 1997; 78: 451–456.
Mazurier C, Goudemand J, Hilbert L, et al. Type 2N von Willebrand disease: clinical manifestations, pathophysiology, laboratory diagnosis and molecular biology. Best Pract Res Clin Haematol 2001; 14: 337–347.
James AH, Ragni MV, Picozzi VJ . Bleeding disorders in premenopausal women: (another) public health crisis for hematology?. Hematol Am Soc Hematol Educ Program 2006; 474–485.
Kadir RA, Chi C . Women and von Willebrand disease: controversies in diagnosis and management. Semin Thromb Hemost 2006; 32: 605–615.
Nurden P, Gobbi G, Nurden A, et al. Abnormal VWF modifies megakaryocytopoiesis: studies of platelets and megakaryocyte cultures from patients with von Willebrand disease type 2B. Blood 2010; 115: 2649–2656.
Casonato A, Pontara E, Sartorello F, et al. Identifying carriers of type 2N von Willebrand disease: procedures and significance. Clin Appl Thromb Hemost 2007; 13: 194–200.
Goodeve AC, Al-Baba S, Panayi M . Mutations leading to reduced von Willebrand factor binding to factor VIII are uncommon among patients referred for type 2N von Willebrand disease genetic analysis. Platelets 2009; 20( S1): S14 O16.
Goodeve A . Molecular genetic testing of hemophilia A. Semin Thromb Hemost 2008; 34: 491–501.
Keeney S, Mitchell M, Goodeve A . The molecular analysis of haemophilia A: a guideline from the UK haemophilia centre doctors' organization haemophilia genetics laboratory network. Haemophilia 2005; 11: 387–397.
Zimmermann MA, Oldenburg J, Muller CR, Rost S . Characterization of duplication breakpoints in the factor VIII gene. J Thromb Haemost 2010; 8: 2696–2704.
Pavlova A, Brondke H, Musebeck J, et al. Molecular mechanisms underlying hemophilia A phenotype in seven females. J Thromb Haemost 2009; 7: 976–982.
Favaloro EJ, Patterson D, Denholm A, et al. Differential identification of a rare form of platelet-type (pseudo-) von Willebrand disease (VWD) from Type 2B VWD using a simplified ristocetin-induced-platelet-agglutination mixing assay and confirmed by genetic analysis. Br J Haematol 2007; 139: 623–626.
Giannini S, Cecchetti L, Mezzasoma AM, Gresele P . Diagnosis of platelet-type von Willebrand disease by flow cytometry. Haematologica 2010; 95: 1021–1024.
Othman M, Favaloro EJ . Genetics of type 2B von Willebrand disease: “true 2B,” “tricky 2B,” or “not 2B.” What are the modifiers of the phenotype?. Semin Thromb Hemost 2008; 34: 520–531.
PT-VWD-Registry. Registry on platelet type von Willebrand disease. Available at: http://www.pt-vwd.org/. Accessed January 10, 2011 .
HGVS. Nomenclature for the description of sequence variations homepage. Available at: http://www.hgvs.org/mutnomen/. Accessed January 10, 2011.
Enayat MS, Guilliatt AM, Lester W, et al. Distinguishing between type 2B and pseudo-von Willebrand disease and its clinical importance. Br J Haematol 2006; 133: 664–666.
Sucker C, Michiels JJ, Zotz RB . Causes, etiology and diagnosis of acquired von Willebrand disease: a prospective diagnostic workup to establish the most effective therapeutic strategies. Acta Haematol 2009; 121: 177–182.
Nichols WL, Rick ME, Ortel TL, et al. Clinical and laboratory diagnosis of von Willebrand disease: a synopsis of the 2008 NHLBI/NIH guidelines. Am J Hematol 2009; 84: 366–370.
Federici AB . Acquired von Willebrand syndrome: an underdiagnosed and misdiagnosed bleeding complication in patients with lymphoproliferative and myeloproliferative disorders. Semin Hematol 2006; 43: S48–S58.
Biss TT, Blanchette VS, Clark DS, et al. Quantitation of bleeding symptoms in children with von Willebrand disease: use of a standardized pediatric bleeding questionnaire. J Thromb Haemost 2010; 8: 950–956.
Demers C, Derzko C, David M, Douglas J . Gynaecological and obstetric management of women with inherited bleeding disorders. J Obstet Gynaecol Can 2005; 27: 707–732.
Castaman G, Lethagen S, Federici AB, et al. Response to desmopressin is influenced by the genotype and phenotype in type 1 von Willebrand disease (VWD): results from the European Study MCMDM-1VWD. Blood 2008; 111: 3531–3539.
Federici AB . The use of desmopressin in von Willebrand disease: the experience of the first 30 years (1977–2007). Haemophilia 2008; 14: suppl 1 5–14.
Federici AB . Highly purified VWF/FVIII concentrates in the treatment and prophylaxis of von Willebrand disease: the PRO.WILL Study. Haemophilia 2007; 13: suppl 5 15–24.
Greaves M, Watson HG . Approach to the diagnosis and management of mild bleeding disorders. J Thromb Haemost 2007; 5: suppl 1 167–174.
ACOG Committee Opinion no. 451 Von Willebrand disease in women. Obstet Gynecol 2009; 114: 1439–1443.
Davis A, Godwin A, Lippman J, Olson W, Kafrissen M . Triphasic norgestimate-ethinyl estradiol for treating dysfunctional uterine bleeding. Obstet Gynecol 2000; 96: 913–920.
Rubin G, Wortman M, Kouides PA . Endometrial ablation for von Willebrand disease-related menorrhagia—experience with seven cases. Haemophilia 2004; 10: 477–482.
James AH, Kouides PA, Abdul-Kadir R, et al. Von Willebrand disease and other bleeding disorders in women: consensus on diagnosis and management from an international expert panel. Am J Obstet Gynecol 2009; 201: 12 e1–e8.
Castaman G, Tosetto A, Rodeghiero F . Pregnancy and delivery in women with von Willebrand's disease and different von Willebrand factor mutations. Haematologica 2010; 95: 963–969.
Varughese J, Cohen AJ . Experience with epidural anaesthesia in pregnant women with von Willebrand disease. Haemophilia 2007; 13: 730–733.
Berntorp E . Prophylaxis in von Willebrand disease. Haemophilia 2008; 14: suppl 5 47–53.
Berntorp E, Windyga J . Treatment and prevention of acute bleedings in von Willebrand disease—efficacy and safety of Wilate, a new generation von Willebrand factor/factor VIII concentrate. Haemophilia 2009; 15: 122–130.
Goodeve A, Castaman G, Rodeghiero F, et al. Rate of de novo VWF mutations in patients historically diagnosed with type 1 VWD in the European study, molecular and clinical markers for the diagnosis and management of type 1 VWD (MCMDM-1VWD). J Thromb Haemost 2007; 5: s2 P-T-189.
Castaman G, Rodeghiero F, Tosetto A, et al. Hemorrhagic symptoms and bleeding risk in obligatory carriers of type 3 von Willebrand disease: an international, multicenter study. J Thromb Haemost 2006; 4: 2164–2169.
Laurie AD, Hill AM, Harraway JR, et al. Preimplantation genetic diagnosis for hemophilia A using indirect linkage analysis and direct genotyping approaches. J Thromb Haemost 2010; 8: 783–789.
El-Toukhy T, Bickerstaff H, Meller S . Preimplantation genetic diagnosis for haematologic conditions. Curr Opin Pediatr 2010; 22: 28–34.
Schneppenheim R, Budde U, Beutel K, et al. Response to DDAVP in children with von Willebrand disease type 2. Hamostaseologie 2009; 29: 143–148.
Sadler JE, Mannucci PM, Berntorp E, et al. Impact, diagnosis and treatment of von Willebrand disease. Thromb Haemost 2000; 84: 160–174.
Mannucci PM, Federici AB . Antibodies to von Willebrand factor in von Willebrand disease. Adv Exp Med Biol 1995; 386: 87–92.
Jenkins PV, O'Donnell JS . ABO blood group determines plasma von Willebrand factor levels: a biologic function after all?. Transfusion 2006; 46: 1836–1844.
Gill JC, Endres-Brooks J, Bauer PJ, Marks WJ, Montgomery RR . The effect of ABO blood group on the diagnosis of von Willebrand disease. Blood 1987; 69: 1691–1695.
Davies JA, Collins PW, Hathaway LS, Bowen DJ . Effect of von Willebrand factor Y/C1584 on in vivo protein level and function and interaction with ABO blood group. Blood 2007; 109: 2840–2846.
Mazurier C, Rodeghiero F . Recommended abbreviations for von Willebrand factor and its activities. Thromb Haemost 2001; 86: 712.
Acknowledgements
Supported by the Zimmerman NIH Program for the “Molecular and Clinical Biology of VWD” (P01 HL081588), and the Bayer Hemophilia Awards Program Early Career Investigator Award (P.D.J.).
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
James, P., Goodeve, A. von Willebrand disease. Genet Med 13, 365–376 (2011). https://doi.org/10.1097/GIM.0b013e3182035931
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e3182035931
Keywords
This article is cited by
-
Vonicog alfa for the management of von Willebrand disease: a comprehensive review and single-center experience
Journal of Thrombosis and Thrombolysis (2020)
-
De novo mutation and somatic mosaicism of gene mutation in type 2A, 2B and 2M VWD
Thrombosis Journal (2016)
-
Analysis of Ebola virus polymerase domains to find strain-specific differences and to gain insight on their pathogenicity
VirusDisease (2016)
-
Salvage therapy with high dose Intravenous Immunoglobulins in acquired Von Willebrand Syndrome and unresponsive severe intestinal bleeding
Experimental Hematology & Oncology (2014)
