
Abstract
Purpose:
The purpose of this study is to describe recessive alleles in strictly dominant genes. Identifying recessive mutations in genes for which only dominant disease or risk alleles have been reported can expand our understanding of the medical relevance of these genes both phenotypically and mechanistically. The Saudi population is enriched for autozygosity, which enhances the homozygous occurrence of alleles, including pathogenic alleles in genes that have been associated only with a dominant inheritance pattern.
Methods:
Exome sequencing of patients from consanguineous families with likely recessive phenotypes was performed. In one family, the genotype of the deceased children was inferred from their parents due to lack of available samples.
Results:
We describe the identification of 11 recessive variants (5 of which are reported here for the first time) in 11 genes for which only dominant disease or risk alleles have been reported. The observed phenotypes for these recessive variants were novel (e.g., FBN2-related myopathy and CSF1R-related brain malformation and osteopetrosis), typical (e.g., ACTG2-related visceral myopathy), or an apparently healthy state (e.g., PDE11A), consistent with the corresponding mouse knockout phenotypes.
Conclusion:
Our results show that, in the era of genomic sequencing and “reverse phenotyping,” recessive variants in dominant genes should not be dismissed based on perceived “incompatibility” with the patient’s phenotype before careful consideration.
Genet Med advance online publication 06 April 2017
Similar content being viewed by others

Introduction
Mendelian genes are so labeled because of the capacity of variants therein to express themselves as discernible phenotypes that follow one of the classical modes of Mendelian inheritance.1 It may be more accurate to refer to variants rather than genes as Mendelian because a given gene can harbor variants that cause Mendelian phenotypes as well as variants that contribute risk to multifactorial traits.2 The power of Mendelian variants to unravel important physiological functions of genes at the organismal level is well known.1
The molecular pathogenesis of Mendelian variants varies widely. Dominant variants can exert their effect by virtue of deficiency of the mutant protein (haploinsufficiency), interference with the normal protein (dominant negative), acquisition of a novel toxic function (toxic gain of function), or loss of the capacity for homeostatic regulation (dominant gain of function). With the exception of the first mechanism, these mechanisms are typically conferred by a highly specific class of variants that can provide valuable insights into the functional topology of proteins, although they may not necessarily reflect the normal physiological roles of these proteins. Loss-of-function variants, however, tend to more faithfully unravel the physiological context of the respective genes. Discerning the exact molecular pathomechanism of dominant variants can be difficult. By contrast, recessive mutations are typically loss-of-function in nature (partial or complete), thus facilitating comparison of the human phenotype with the corresponding knockout animal models.
The abovementioned differences between dominant and recessive mutations have important clinical and basic scientific implications. For example, dominant and recessive mutations in the same gene can express themselves in markedly different phenotypes. Conversely, when the dominant and recessive phenotypes are distinctly different, it can be assumed that the molecular pathomechanism of the dominant mutations is not simple haploinsufficiency. This is important in the context of molecular karyotyping because the recommendation of the American College of Medical Genetics and Genomics (ACMG) is to focus on dominant disease genes with established haploinsufficiency as the molecular pathomechanism when reporting the gene content of hemizygous deletions.3 Furthermore, the requirement for efficient filters to handle the massive number of variants identified by genomic sequencing can risk eliminating causal recessive variants in genes in which only dominant alleles have been reported when the reported phenotype shows little or no resemblance to the patient’s presentation.
In our search for novel genotype–phenotype correlations, we have published many novel disease and candidate disease genes.4 However, we have also encountered a number of known dominant genes for which the observed recessive phenotype was surprisingly different.5 Examples include ELOV4-related Sjögren-Larssen syndrome vs. Stargardt macular degeneration, BRCA2-related severe microcephalic primordial dwarfism vs. hereditary breast and ovarian cancer, and APC-related Cenani-Lenz syndrome vs. hereditary adenomatous polyposis coli.6,7,8 In this study, we report additional novel recessive mutations in known dominant diseases and discuss the biological and medical relevance of this information to the application of clinical genomics.
Materials and Methods
Human subjects
All patients described in this study were enrolled with informed consent as part of various institutional review board–approved protocols, depending on the clinical presentation (KFSHRC RAC 2080006, 2070023, and 2121053).
Identification of recessive mutations
Cases typically undergo autozygome analysis using the Axiom SNP Chip (Affymetrix, Santa Clara, CA) according to the manufacturer’s instructions, followed by mapping of runs of homozygosity >2 Mb as surrogates of autozygosity using AutoSNPa. When multiple affected individuals were available, we searched for their overlapping autozygome, as described in detail elsewhere.9 When the index was unavailable, a search for the likely disease haplotype was conducted by comparing the parents using IBDelphi.10 Exome sequencing was typically performed only on the index; when the index was unavailable, we resorted to exome sequencing of both parents to infer the genotype of the index. Exome variants were filtered by the coordinates of the candidate autozygome or, in the case of parents with no available affected children, the candidate haplotypes, as determined by AutoSNPa and IBDelphi, respectively.11 We only considered novel or very rare (minor allele frequency <0.001) variants based on an in-house database of 2,793 Saudi exomes (from patients with various Mendelian diseases) and ExAC that were either coding or in the flanking intronic sequences. Because GABRD and FBN2 were also included in the Neurology and Dysmorphology and Skeletal Dysplasia gene panels, respectively, an additional 1,787 and 739 Saudis were screened for these two genes, respectively. We considered the candidacy of genes even if the published phenotype appeared inconsistent with the patient’s presentation. Sanger confirmation was performed for variants discussed in this article, as was segregation analysis for all available members, which was consistent with their candidacy as fully penetrant autosomal recessive variants. We included in this study only autosomal recessive mutations in genes that had been linked to human phenotypes in a strictly autosomal dominant fashion.
Computational structural analysis of mutants
Sequences were retrieved from the UniProt database. RaptorX was used for prediction of secondary structures and protein disorder, and RaptorX and SWISS-MODEL were used to produce homology models.12,13 Models were manually inspected, and mutations were evaluated using the PyMOL program (http://pymol.org). Transmembrane helices were predicted using Phobius.14 Functional information was compiled from various resources, including UniProt, InterPro,15 and publications associated with the model templates used.
Results
We identified a total of 11 likely causal autosomal recessive variants in genes that had only been linked to human phenotypes in an autosomal dominant fashion, five of which are described here for the first time (pedigrees are shown in Supplementary Figure S1 online). Unless stated otherwise, the heterozygous parents and siblings were asymptomatic. These variants fall into three categories and are described in detail below.
Recessive variants that cause similar phenotypes to the dominant phenotypes
We report the first homozygous truncating mutation in ACTG2 in a 16-year-old girl with severe visceral myopathy. Although this is the same phenotypic category of the previously established ACTG2-related dominant phenotype, we note that the phenotype is very severe; it caused death in the sister (24 years old at the time of death) due to intestinal obstruction and perforation (presumed to be homozygous but never tested) and similarly severe recurrent intestinal obstruction requiring intestinal transplant in the index case ( Figure 1 ). The heterozygous parents (confirmed in father and presumed in mother who refused testing) and siblings reported chronic intermittent constipation, suggesting a semidominant inheritance. Our finding is consistent with the previously suggested haploinsufficiency mechanism.16 Interestingly, an adult sister with a history of chronic constipation similar to that of the parents and heterozygous siblings was found to be homozygous, which suggests that the severe intestinal phenotype observed in the living homozygous and presumed homozygous deceased sisters was incompletely penetrant (Supplementary Figure S1 online).

Recessive mutation in ACTG2 in family 15DG1406. (a) Plain abdominal radiograph showing significant distention of the colon. (b) Water-soluble contrast enema showing distended rectum, sigmoid, and descending colon and obstruction suggestive of transverse colon volvulus.
A heterozygous variant in GABRD, specifically Glu177Ala, has been reported to contribute to epilepsy at reduced penetrance in one family.17 Although a homozygous Arg220His was also reported in the context of epilepsy, the high frequency of this variant in ExAC is incompatible with being disease-causing. In 16NGS-0186, a 24-month-old boy who presented with global developmental delay, severe hypotonia, and epileptic encephalopathy, we identified a novel homozygous missense variant in GABRD: NM_000815.4:c.875C>T:p.(Thr292Met). According to ACMG guidelines,18 this variant should be classified as variants of unknown significance because the only support of potential pathogenicity comes from its complete absence in controls, its segregation with the disease within the family, and in silico prediction (PolyPhen (1), SIFT (sorting intolerant from tolerant) (0), and CADD (combined annotation-dependent depletion) (18)). Thr292 is located in the membrane-integral channel part of the protein (which consists of an extracellular ligand-binding domain and a membrane-spanning channel domain) (Supplementary Figure S2 online). In the pentamer, Thr292 is located at the inner side of the channel. The replacement of the short, rigid, and partly hydrophilic threonine by a long flexible and completely hydrophobic methionine is expected to substantially alter the inner side of the channel and hence the passage of molecules (Supplementary Figure S2 online). Pathogenicity is further substantiated by the recently described pLI (probability of being loss of function–intolerant) score of 0.92 for GABRD19 as well as the phenotype of Gabrd-/- mice that have increased seizure susceptibility.20 It remains to be seen, through the identification of future patients, whether recessive GABRD variants are a bona fide cause of epileptic encephalopathy.
Recessive mutations with distinctly different phenotypes compared with the dominant phenotypes
Case 16DG0107 presented to us at the age of 13.5 years because of motor difficulties. Bilateral clubfoot deformity was noted immediately after birth and was managed with serial casting. Although walking was not particularly delayed (17 m), his gait has always been abnormal and his clubfoot deformity slowly recurred. He had serious functional limitations at the time of presentation, when he could walk only with difficulty and could not run. He fatigues easily and experiences dyspnea on exertion despite a normal cardiac workup. He has a history of frequent patellar dislocations, for which he receives supportive therapy. Magnetic resonance imaging of the spine revealed mild L4/L5 and L5/S1 disc herniation and abnormal bulging of the discs in the cervical spine, with no evidence of spinal canal stenosis or cord compression. His neurosurgical evaluation is ongoing. He has gynecomastia, which is being investigated by endocrinology, but to date the workup has been negative. Physical examination revealed he was overweight (height: 168.5 cm; weight: 80 kg) and macrocephalic (occipitofrontal circumference 59 cm, +3.2 SD); he had a square face, pointy chin, camptodactyly with underdeveloped or absent interphalangeal (especially distal) creases, prominent skin striae ( Figure 2a , b ), limited knee extension, and abnormal gait with significant lordosis, camptocormia, flexed knees, and intoeing. His motor examination revealed severe muscle weakness in the hip extensors (gluteus maximus) and paraspinal muscles, with mild deltoid, iliopsoas, quadriceps, and hamstring involvement. The serum creatine kinase level was normal. Magnetic resonance imaging revealed gluteus maximus fatty infiltration.

Recessive mutation in FBN2 in family 16DG0107. (a) and (b) Representative images of the widespread striae. (c) The muscle biopsy results show myopathic features with variation in fiber size and scattered atrophic fibers (arrow, hematoxylin and eosin, ×400). (d) Ultrastructural examination shows scattered foci of myofibrillar disruption (arrow, EM ×4,000). (e) Rodlike structures are noted occasionally at the periphery of some fibers, within a background of myofibrillar disarray (EM ×25,000). (f) Computational 3D models of FBN2 residues 1–28 (which constitute the signal peptide) for the wild-type Leu14 (left) and mutant Arg14 (right). Position 14 is encircled. Orientation is with the N-terminal upward. Top panels: ribbon representation. Bottom panels: electrostatic surface, color-ramped from blue (basic/positively charged) to gray (uncharged/hydrophobic) to red (acidic/negatively charged). The theoretical 3D model has been established based on a helical secondary structure prediction for residues 2–28 and weak homology to PDB id 5i6c (P value = 3.7 × 10−2).
Muscle biopsy showed unusual myofibrillar disorganization; there was variation in fiber size, with readily identifiable atrophic fibers, primarily type I, and EM showed a generalized disarray of fibers ( Figure 2c – e ). Exome sequencing revealed a novel homozygous FBN2 variant NM_001999.3:c.41T>G:p.(Leu14Arg) that was homozygous only in him, not in his two siblings, whereas the parents were heterozygous for the change. This was the only novel coding/splicing variant in the index’s exome, and it was predicted to be pathogenic by PolyPhen (0.67), SIFT (0.00), and CADD (18). Leucine 14 is located in a 110-residue N-terminal fragment prior to the first epidermal growth factor–like domain. This fragment is predicted to be unstructured (RaptorX disorder probability >90%, except for residues 86–103, where disorder probability is 70–90%). Leu14 is located in a highly hydrophobic sequence (LYFLWL) that follows after an N-terminal basic region ( Figure 2f ). These N-terminal 28 residues are predicted to be a signal peptide required for the correct secretion of the protein (Phobius posterior probability of 1). The nonhomologous substitution of the medium-size hydrophobic leucine with a long, positively charged arginine disrupts this recognition region ( Figure 2f ). It is therefore expected that the Leu14Arg mutation significantly affects correct secretion of the mutant, leading to abnormal FBN2 protein levels in the extracellular microfibrils. Expected effects include abnormal elasticity and extension of elastic fibers and developmental phenotypes. Based on our analysis, we noted that the severe muscle involvement in Fbn2-/- was highly consistent with the hypomorphic/nullimorphic nature of the Leu14Arg.21 Furthermore, the only human phenotype linked to FBN2 is a dominant congenital form of contractural arachnodactyly; the clustering of these dominant variants in specific domains in FBN2 is consistent with a mechanism other than haploinsufficiency for the dominant disease.22,23,24
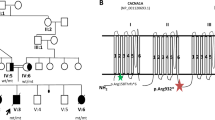
16DG0825 and 16DG0826 were a first-cousin couple who lost two children with a lethal phenotype that involves severe brain malformation and generalized osteopetrosis, but no specific diagnosis was reached ( Figure 3 ). Brain imaging was consistent in both children and showed Dandy-Walker malformation, supratentorial ventriculomegaly that was mainly occipital (colpocephaly), agenesis of the corpus callosum, and diffuse bilateral subependymal and periventricular extensive linear calcifications. Intraventricular and subdural hemorrhage as well as thinning of the cortical mantle with poor white/gray matter differentiation and poor gyration were also noted.

Recessive mutation in CSF1R in families 16DG0825 and 16DG0826. (a) Generalized increased bone density with bone sclerotic appearance suggesting osteopetrosis. (b) Dandy-Walker malformation comprising large posterior fossa, a high position of the tentorium cerebelli, hypoplastic vermis, and cerebellum with cystic dilatation of the fourth ventricle widely communicating with a posterior fossa cyst filling nearly the entire posterior fossa.
Owing to the lack of available samples from either of the deceased children, we resorted to “linkage by exclusion”; i.e., we exome-sequenced both parents and filtered the exomic variants by the coordinates of haplotypes that were not observed in the autozygous state in any of their four healthy children. Only one likely pathogenic variant was identified using this method: a novel truncating variant in CSF1R (NM_001288705.1:c.1620T>A:p.(Tyr540*)) that was heterozygous in the parents and either absent or heterozygous in their four children. Heterozygous changes in CSF1R are known to cause an adult form of encephalomyopathy, presumably via a dominant negative effect of the mutant copy on the wild-type copy because this receptor functions via ligand-dependent dimerization and autophosphorylation.25,26 Unlike previously reported dominant mutations, our mutation completely abolishes the intracellular part of the receptor, which starts at amino acid 537. As such, the mutant form cannot exert a dominant negative effect and will most likely display haploinsufficiency, which we showed in the heterozygotes, including the adult parents, to be benign. Because CSF1R works by ligand-dependent dimerization and autophosphorylation, heterozygotes would express only wild-type CSF1R on the cell surface, which should function normally. More importantly, mice deficient for CSF1R recapitulated the osteopetrosis, brain involvement, and early lethality observed in the two children.27,28 However, because homozygosity for the truncating CSF1R mutation in these children was not directly demonstrated, this scenario remains hypothetical.
Homozygous truncating variants in dominant genes with no apparent phenotype
Case 16DG0991 was an infant who died at 10 months of age as a result of complications from unexplained persistent cholestasis and bilateral dysplastic kidneys. Exome sequencing revealed a homozygous truncating variant in PDE11A: NM_016953.3:c.1633C>T:p.(Arg545*). However, the same variant was identified in a healthy 6-year-old sibling. PDE11A heterozygous missense variants have been reported in patients with adrenocortical tumors.29 However, we noted normal adrenocortical structure and function in the index and her healthy sibling, both of whom had presumed complete loss of function of PDE11A. This observation, the very low PLI score of PDE11A (0.00),19 and the occurrence of three cases in ExAC with homozygous truncating mutations in PDE11A strongly suggest that the gene may be dispensable for adrenocortical function and that the reported tumor phenotype was caused by a gain-of-function mechanism. Interestingly, the same variant we observed in this family (Arg545*) was also reported in the context of a testicular germ cell tumor.30 However, there was no history of such a tumor in either the carrier father or the extended pedigree.
Discussion
Variant interpretation remains the most formidable challenge in the delivery of clinical genomics. An important element of variant interpretation is knowledge of previously reported phenotypes, not necessarily for the variant in question but for the gene itself. Allelic disorders are characterized by apparent phenotypic disparity despite being caused by different alleles in the same gene. The mechanisms that underlie allelism vary. Although some allelic disorders simply represent a severity spectrum of the same phenotype, many are distinct. Knowledge of the mechanism underlying allelism can be critical to the interpretation of variants. For example, a novel variant in the gene AR (androgen receptor) with presumed deleterious effects on the protein can be predicted to result in a phenotype related to the endocrinological function of the androgen receptor rather than Kennedy disease because the latter results from a very specific class of variants that confer a novel toxic property on the protein, with resulting neurological deterioration.31
When allelism is observed in the context of multiple modes of inheritance, it can provide an opportunity to understand the true pathomechanism of disease.32 For example, there was debate regarding whether the mechanism of ELOVL4-related autosomal dominant Stargardt disease is dominant gain of function or haploinsufficiency.33 However, our finding that homozygous truncating mutations in ELOVL4 cause a severe autosomal recessive Sjögren-Larssen syndrome while the “haploinsufficient” parents have normal eye examination strongly favors the dominant negative model of Stargardt disease-related variants.6
In this paper, we have presented our experience with recessive variants observed in genes that had been linked to human disease only in the heterozygous state and speculate on the insights that can be gained from such observations by classifying these variants into three categories. In the first category, the recessive phenotype is similar to, but more severe than, the dominant phenotype. For these variants, it can be argued that these are dominant variants with variable penetrance, e.g., the mild constipation in the heterozygous parents and siblings of two girls with a lethal from of ACTG2-related visceral myopathy.
By contrast, variants in the second category—those with a recessive phenotype that is distinctly different from the reported dominant phenotype—appear to lend themselves to potentially novel mechanistic interpretation. The examples we present in this regard require additional studies to establish the clinical significance of the variants and to determine the mode of inheritance. However, we note that the remarkable concordance between the phenotypes we observed in our homozygous patients and the knockout mouse model argues in favor of these being the bona fide autosomal recessive loss-of-function phenotype in humans. This can be readily seen in the case of FBN2-related myopathy vs. myopathy in Fbn2-/-, as well as in CSF1R-related osteopetrosis, brain malformation, and early lethality vs. osteopetrosis and early lethality in Csf1r-/-. Similarly, the apparent lack of adrenocortical phenotype in the two individuals with homozygous truncation of PDE11A was consistent with the phenotype of Pde11a-/-,34 suggesting that adrenocortical tumor–related PDE11A variants probably confer gain of function.
The findings of our study emphasize that deleterious heterozygous variants, especially novel ones, in dominant genes encountered among “healthy” individuals may not necessarily represent instances of reduced penetrance but, rather, true instances of autosomal recessive inheritance. This can have a significant impact on the way these variants are reported and on the genetic counseling of these individuals. Finally, the apparent disparity between dominant and recessive phenotypes for some genes should serve as a reminder that variants should not be dismissed as potential causes of phenotypes solely on the basis of the perceived incompatibility of the patient’s phenotype with what has previously been linked to the gene in question.
In conclusion, we add to the growing list of genes that can cause phenotypes in both dominant and recessive states. We show that the power of autozygosity to enhance the occurrence of homozygous variants provides a valuable resource for a deeper understanding of the phenotypic spectrum of not only recessive but also dominant genes.
Disclosure
The authors declare no conflict of interest.
References
Alkuraya FS. Discovery of mutations for Mendelian disorders. Hum Genet 2016;135:615–623.
Alkuraya FS. Human knockout research: new horizons and opportunities. Trends Genet 2015;31:108–115.
Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST ; Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med 2011;13:680–685.
Alkuraya FS. Genetics and genomic medicine in Saudi Arabia. Mol Genet Genomic Med 2014;2:369–378.
Shamia A, Shaheen R, Sabbagh N, Almoisheer A, Halees A, Alkuraya FS. Revisiting disease genes based on whole-exome sequencing in consanguineous populations. Hum Genet 2015;134:1029–1034.
Aldahmesh MA, Mohamed JY, Alkuraya HS, et al. Recessive mutations in ELOVL4 cause ichthyosis, intellectual disability, and spastic quadriplegia. Am J Hum Genet 2011;89:745–750.
Shaheen R, Faqeih E, Ansari S, et al. Genomic analysis of primordial dwarfism reveals novel disease genes. Genome Res 2014;24:291–299.
Patel N, Faqeih E, Anazi S, et al. A novel APC mutation defines a second locus for Cenani–Lenz syndrome. J Med Genet 2015;52:317–321.
Alkuraya FS. Discovery of rare homozygous mutations from studies of consanguineous pedigrees. Curr Protoc Hum Genet 2012; ch 6, unit 6.12.
Carr IM, Markham SA, Pena SD. Estimating the degree of identity by descent in consanguineous couples. Hum Mutat 2011;32:1350–1358.
Alkuraya FS. The application of next-generation sequencing in the autozygosity mapping of human recessive diseases. Hum Genet 2013;132:1197–1211.
Källberg M, Margaryan G, Wang S, Ma J, Xu J. RaptorX server: a resource for template-based protein structure modeling. Methods Mol Biol 2014;1137:17–27.
Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 2006;22:195–201.
Käll L, Krogh A, Sonnhammer EL. A combined transmembrane topology and signal peptide prediction method. J Mol Biol 2004;338:1027–1036.
Hunter S, Jones P, Mitchell A, et al. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res 2012 40(Database issue):D306-12.
Wangler MF, Beaudet AL. ACTG2-related disorders. In: Pagon RA, Adam MP, Ardinger HH, et al. (eds). GeneReviews. University of Washington: Seattle, WA , 1993.
Dibbens LM, Feng HJ, Richards MC, et al. GABRD encoding a protein for extra- or peri-synaptic GABAA receptors is a susceptibility locus for generalized epilepsies. Hum Mol Genet 2004;13:1315–1319.
Richards S, Aziz N, Bale S, et al.; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424.
Lek M, Karczewski KJ, Minikel EV, et al.; Exome Aggregation Consortium. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–291.
Spigelman I, Li Z, Banerjee PK, Mihalek RM, Homanics GE, Olsen RW. Behavior and physiology of mice lacking the GABAA-receptor delta subunit. Epilepsia 2002;43(suppl 5):3–8.
Sengle G, Carlberg V, Tufa SF, et al. Abnormal activation of BMP signaling causes myopathy in Fbn2 null mice. PLoS Genet 2015;11:e1005340.
Park ES, Putnam EA, Chitayat D, Child A, Milewicz DM. Clustering of FBN2 mutations in patients with congenital contractural arachnodactyly indicates an important role of the domains encoded by exons 24 through 34 during human development. Am J Med Genet 1998;78:350–355.
Putnam EA, Zhang H, Ramirez F, Milewicz DM. Fibrillin-2 (FBN2) mutations result in the Marfan-like disorder, congenital contractural arachnodactyly. Nat Genet 1995;11:456–458.
Gupta PA, Putnam EA, Carmical SG, et al. Ten novel FBN2 mutations in congenital contractural arachnodactyly: delineation of the molecular pathogenesis and clinical phenotype. Hum Mutat 2002;19:39–48.
Rademakers R, Baker M, Nicholson AM, et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet 2011;44:200–205.
Pridans C, Sauter KA, Baer K, Kissel H, Hume DA. CSF1R mutations in hereditary diffuse leukoencephalopathy with spheroids are loss of function. Sci Rep 2013;3:3013.
Dai XM, Ryan GR, Hapel AJ, et al. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood 2002;99:111–120.
Li J, Chen K, Zhu L, Pollard JW. Conditional deletion of the colony stimulating factor-1 receptor (c-fms proto-oncogene) in mice. Genesis 2006;44:328–335.
Horvath A, Boikos S, Giatzakis C, et al. A genome-wide scan identifies mutations in the gene encoding phosphodiesterase 11A4 (PDE11A) in individuals with adrenocortical hyperplasia. Nat Genet 2006;38:794–800.
Pathak A, Stewart DR, Faucz FR, et al. Rare inactivating PDE11A variants associated with testicular germ cell tumors. Endocr Relat Cancer 2015;22:909–917.
Fischbeck KH. Kennedy disease. J Inherit Metab Dis 1997;20:152–158.
Zschocke J. Dominant versus recessive: molecular mechanisms in metabolic disease. J Inherit Metab Dis 2008;31:599–618.
Grayson C, Molday RS. Dominant negative mechanism underlies autosomal dominant Stargardt-like macular dystrophy linked to mutations in ELOVL4. J Biol Chem 2005;280:32521–32530.
Kelly MP, Logue SF, Brennan J, et al. Phosphodiesterase 11A in brain is enriched in ventral hippocampus and deletion causes psychiatric disease-related phenotypes. Proc Natl Acad Sci 2010;107:8457–8462.
Karaca E, Harel T, Pehlivan D, et al. Genes that affect brain structure and function identified by rare variant analyses of Mendelian neurologic disease. Neuron 2015;88:499–513.
Acknowledgements
We thank the study families for their enthusiastic participation. We also thank the Sequencing and Genotyping Core Facilities at KFSHRC for their technical help. This work was supported by a King Salman Center for Disability Research grant (F.S.A.) as well as KACST grant 15-BIO3688-20 (F.S.A.). The research by S.T.A. reported in this publication was supported by funding from King Abdullah University of Science and Technology (KAUST).
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Figures
(PDF 502 kb)
Rights and permissions
About this article

Cite this article
Monies, D., Maddirevula, S., Kurdi, W. et al. Autozygosity reveals recessive mutations and novel mechanisms in dominant genes: implications in variant interpretation. Genet Med 19, 1144–1150 (2017). https://doi.org/10.1038/gim.2017.22
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2017.22
Keywords
This article is cited by
-
Clinical application of next generation sequencing for Mendelian disease diagnosis in the Iranian population
npj Genomic Medicine (2024)
-
Brain abnormalities, neurodegeneration, and dysosteosclerosis (BANDDOS): new cases, systematic literature review, and associations with CSF1R-ALSP
Orphanet Journal of Rare Diseases (2023)
-
Tissue-specific macrophages: how they develop and choreograph tissue biology
Nature Reviews Immunology (2023)
-
The Role of Tumor Epithelial-Mesenchymal Transition and Macrophage Crosstalk in Cancer Progression
Current Osteoporosis Reports (2023)
-
Diagnostic implications of pitfalls in causal variant identification based on 4577 molecularly characterized families
Nature Communications (2023)
