
Abstract
Purpose
The clinical utility of screening unselected individuals for pathogenic BRCA1/2 variants has not been established. Data on cancer risk management behaviors and diagnoses of BRCA1/2-associated cancers can help inform assessments of clinical utility.
Methods
Whole-exome sequences of participants in the MyCode Community Health Initiative were reviewed for pathogenic/likely pathogenic BRCA1/2 variants. Clinically confirmed variants were disclosed to patient–participants and their clinicians. We queried patient–participants’ electronic health records for BRCA1/2-associated cancer diagnoses and risk management that occurred within 12 months after results disclosure, and calculated the percentage of patient–participants of eligible age who had begun risk management.
Results
Thirty-seven MyCode patient–participants were unaware of their pathogenic/likely pathogenic BRCA1/2 variant, had not had a BRCA1/2-associated cancer, and had 12 months of follow-up. Of the 33 who were of an age to begin BRCA1/2-associated risk management, 26 (79%) had performed at least one such procedure. Three were diagnosed with an early-stage, BRCA1/2-associated cancer—including a stage 1C fallopian tube cancer—via these procedures.
Conclusion
Screening for pathogenic BRCA1/2 variants among unselected individuals can lead to occult cancer detection shortly after disclosure. Comprehensive outcomes data generated within our learning healthcare system will aid in determining whether population-wide BRCA1/2 genomic screening programs offer clinical utility.
Similar content being viewed by others


Introduction
The clinical utility of screening unselected individuals for pathogenic BRCA1/2 variants has not been established. Proponents point to (i) the well-established potential for reducing morbidity and mortality in individuals found to have a pathogenic BRCA1/2 variant1 and (ii) the significant proportion of the population with a cancer family history that would not prompt clinical attention.2 Others caution that implementing population BRCA1/2 screening is premature without evidence of clinical utility among unselected individuals.3 BRCA1/2-associated morbidity and mortality data4 are derived from individuals identified because of a personal or family history that raised suspicion of a BRCA1/2 mutation (i.e., indication-based testing). If these benefits do not emerge from genomic screening of unselected individuals, the associated medical, psychosocial, and financial costs of surveillance and prophylactic surgery might not be justified.3
Determining the clinical utility of a BRCA1/2 screening program in unselected individuals requires longitudinal data on (i) prevalence and penetrance of pathogenic BRCA1/2 variants in diverse, unselected populations; (ii) impact of risk management on cancer morbidity and mortality; (iii) adherence to recommended risk management in the absence of compelling family cancer history; (iv) costs associated with risk management procedures; and (v) psychological outcomes of receiving a pathogenic BRCA variant via the screening program.3 Here we report two early measures of the impact of a BRCA1/2 screening program among individuals previously unaware that they were carrying a pathogenic variant–risk-management initiation and cancer diagnoses prompted by results disclosure.
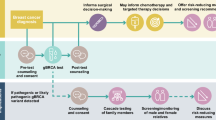
This assessment was performed through the Geisinger Health System (GHS) MyCode Community Health Initiative (hereafter “MyCode”), a research project in which participants’ whole exome sequences are reviewed for pathogenic and likely pathogenic variants in 76 genes (including BRCA1/2 and 54 genes originally proposed by the American College of Medical Genetics and Genomics5) associated with 27 medically actionable conditions. Clinically confirmed variants are disclosed to patient–participants and their clinicians, who are supported in integrating results into care.
Materials and methods
MyCode enrollment process and targeted screening for genomic findings
GHS established MyCode in 2007 as a discovery research initiative enabled by a biobank. Recruitment occurs in primary-care and specialty clinics throughout GHS without regard to underlying disease or cancer risk, and without specific interest in genomic testing. In 2014 GHS established a research collaboration with Regeneron Genetics Center (RGC) that includes conducting whole-exome sequencing in MyCode participants and linking sequence data to participants’ electronic health record (EHR) data. After significant engagement with patient–partners, ethicists, and clinicians, GHS developed an institutional review board–approved protocol (GenomeFIRST) to assess MyCode participants’ exomes for medically actionable findings in 76 genes (including BRCA1/2), confirm results in a Clinical Laboratory Improvement Amendments–certified clinical diagnostic laboratory (Laboratory for Molecular Medicine, LMM), return clinically confirmed results to patient–participants and clinicians, initiate guidelines-based risk assessment and management, and facilitate cascade testing for at-risk family members.6
Whole-exome sequencing and variant classification
Whole-exome sequencing is performed at RGC as described elsewhere.7 Independent teams at GHS, RGC, and the LMM perform bioinformatics reviews of BRCA1/2 variant call files and agree to consensus classification as pathogenic or likely pathogenic according to American College of Medical Genetics and Genomics criteria.8 An independent DNA sample is sent to the LMM for Sanger confirmation of the variants. After this confirmation the LMM sends a clinical laboratory report to the Geisinger Clinical Genomics team.
Clinical program for returning genomic results
In the cases described in this brief report, the Geisinger Clinical Genomics team (genetic counselors, medical geneticists, a nurse practitioner, and administrative staff) received the clinical laboratory report of the pathogenic variant and initiated a disclosure and follow-up protocol. After Clinical Genomics notified the primary-care physician (PCP) and patient–participant, Geisinger’s multidisciplinary Inherited Risk Breast Clinic (genetic counselor, breast surgeon, physician assistant, breast oncologist, and clinical psychologist), Clinical Genomics, and PCPs coordinated medical evaluation9 and cancer risk management. Risk management recommendations were based on National Comprehensive Cancer Network guidelines (which include serial sectioning of fallopian tubes following risk-reducing salpingo-oophorectomy, and prostate cancer surveillance for men with a pathogenic BRCA2 variant).10
Tracking clinical outcomes
The Clinical Genomics team defined outcomes relevant to recommended risk management and cancer diagnoses and tracked them via EHR review. These standardized outcomes (including dates and results of BRCA1/2-associated surveillance and prophylactic surgery and diagnoses of BRCA1/2-associated cancers) were aggregated in a database maintained for all participants with a disclosed result. We chose to assess outcomes at 12 months after disclosure to allow sufficient time for patient–participants to have undergone evaluation and to initiate risk management. For females, mammogram, breast magnetic resonance imaging (MRI), risk-reducing mastectomy, risk-reducing salpingo-oophorectomy, CA125 testing, and transvaginal ultrasound were considered to be BRCA1/2-associated, per National Comprehensive Cancer Network guidelines.10 For males, clinical breast exam and prostate-specific antigen (PSA) testing were considered to be BRCA1/2-associated. We considered melanoma and breast, ovarian, prostate, and pancreatic cancers to be BRCA1/2-associated.
Results
As of 1 April 2017, 55 MyCode participants and their PCPs had been notified of a clinically confirmed pathogenic/likely pathogenic BRCA1/2 variant and followed for at least 12 months. Seven of these were already aware of their BRCA1/2 variant via clinical testing. An additional 11 had a personal history of BRCA1/2-associated cancer. The remaining 37 patient–participants included 17 females (46%) and had a median age of 60 years (mean 56.1 years, range 26–87 years).
Of the 33 patient–participants old enough to perform BRCA1/2-associated risk management, 26 (79%, 15 females, 11 males) had performed at least one such behavior. Three of these patient-participants were diagnosed via these procedures with an early-stage, BRCA1/2-associated cancer, a subclinical phenotype revealed through evaluation.9
The first patient–participant was a 56-year-old woman with a pathogenic BRCA1 variant (ClinVar variation ID 125513). Her medical history was positive for Crohn’s disease and she had had a basal cell carcinoma removed in her 40s, but the history was otherwise negative for cancer. EHR-documented family history (Figure 1a) did not meet guidelines for referral to genetic counseling.10 Her most recent screening mammogram, made at age 52, was normal. Following disclosure she had a normal mammogram, breast MRI, CA125 testing, pelvic ultrasound, and a risk-reducing salpingo-oophorectomy (Figure 2). Final pathology on a 1.4 cm mass first found intraoperatively during the risk-reducing salpingo-oophorectomy revealed a high-grade, serous carcinoma with stromal invasion and a focal serous tubal intraepithelial carcinoma in the right fallopian tube (Supplementary Figure 1a and b online). Peritoneal washes demonstrated malignant cells, indicating stage 1C, which has a 5-year survival of 85%.11

Patient-participants’ family cancer histories. (a–c) Patient-participants’ family cancer histories, as documented in electronic health records prior to BRCA1/2 results disclosure.

Timeline of BRCA1/2 variant disclosure, medical evaluation, risk management, and diagnostic workup (cases 1–3). RRSO, risk-reducing salpingo-oophorectomy; PSA, prostate-specific antigen.
The second patient–participant was a 57-year-old woman with a pathogenic BRCA2 variant (ClinVar Variation ID 9320). Past medical history was significant for a bilateral salpingo-oophorectomy and a hysterectomy at age 42 for fibroids but was negative for cancer. EHR-documented family history (Figure 1b) was notable for a brother who died of pancreatic cancer at 37 and two relatives with colon cancer, but she did not meet referral guidelines for hereditary breast cancer risk.10 Her most recent screening mammogram, at age 53, was normal. Following results disclosure, breast MRI showed a nonmass enhancement in the left breast; subsequent left diagnostic mammogram and targeted second-look ultrasound in the upper outer quadrant were normal (Figure 2; Supplementary Figure 1c–f). MRI-guided biopsy revealed ductal carcinoma in situ (Supplementary Figure 1g). The patient–participant underwent bilateral mastectomy and breast reconstruction. Final pathology showed ductal carcinoma in situ measuring 1.2 cm in its greatest dimension, grade 2 to 3, with negative sentinel lymph nodes—a stage 0 cancer associated with 20-year survival of 97%.12
The third patient–participant was a 55-year-old man with a pathogenic BRCA2 variant (ClinVar Variation ID 38082) and no personal history of cancer. EHR-documented family history (Figure 1c) did not meet genetic counseling referral guidelines.10 Past medical history was significant for morbid obesity, status post Roux-en-Y gastric bypass surgery. Additionally, he had two mildly elevated PSA tests in 2007 (3.27 ng/ml—normal is less than 3.1 ng/ml) and 2008 (3.24 ng/ml) and a markedly elevated PSA test in 2015 (16.60 ng/ml). He was referred to urology after the latter elevated PSA test but did not attend an appointment. Disclosure of his BRCA2 variant prompted his PCP to order another PSA test (eight months after the previous test), which was again markedly elevated, at 19.12 ng/ml (Figure 2). Clinical stage, based on PSA and normal digital rectal exam, was cT1c. A prostate biopsy revealed adenocarcinoma in 5/12 cores (Gleason score 4 + 4 = 8 in one core, 4 + 3 = 7 in four other cores; Supplementary Figure 1h). Computed tomography of abdomen/pelvis and bone scans were negative for metastases.
Discussion
Three of 37 unselected individuals with a pathogenic BRCA1/2 variant detected through a genomic screening program were found to have an early-stage cancer via their initial post-disclosure evaluations and risk management procedures. This is consistent with rates of occult cancer detection upon surveillance and prophylactic surgery in individuals whose BRCA1/2 mutation was identified via family history.13, 14 These cancers were found in individuals without compelling family history documented in the EHR, adding support to the view that genomic screening programs for BRCA1/2 variants can identify at-risk individuals who otherwise would not have come to medical attention.2 By relying on genotypic detection of risk rather than on family history, the BRCA1/2 screening approach focuses on individuals who may derive the greatest benefit from identification of risk.
Finding cancer early is not universally beneficial, as evidenced by concerns about over-diagnosis of cancer and the inability of screening modalities such as PSA testing to distinguish indolent from aggressive cancers.15, 16 Among these three cases, the benefit of early detection is clearest in the patient–participant with an early-stage diagnosis of ovarian cancer and a precursor lesion, which are difficult to screen for effectively.17 Owing in part to these screening limitations, ovarian cancer is detected at a metastatic stage in 60% of cases, with associated 5-year survival of 29%.11
The other two patient–participants might have benefited from genomic screening and subsequent detection of an early cancer, as well. In the patient–participant with ductal carcinoma in situ, the BRCA2 result led to performing breast MRI per the evidence-based guideline10—a test that was otherwise not indicated. This facilitated detection of a mammographically occult, early-stage cancer. That the ductal carcinoma in situ was intermediate- to high-grade is significant, as there is evidence of survival benefit from surgical treatment in such cases.18 In the prostate cancer case, the patient–participant’s PCP used the BRCA1/2 variant to make a risk-based recommendation of a screening test (PSA) of controversial value to the general population.15 As the patient–participant informed the Clinical Genomics team, knowledge of the BRCA2 variant motivated him to follow up an elevated PSA result, which he had not done when prior PSA tests had been elevated. This follow-up led to diagnosis of an aggressive, Gleason 8 prostate cancer.
Our early data on performance of recommended cancer risk management—with 79% of eligible patient–participants having performed some risk management—indicate that genotypically detected variant carriers might place a similar value on risk management as their unaffected counterparts detected via family history.19 The degree to which occult cancer detection and performance of recommended risk management will be replicated in other populations will be influenced by a wide variety of factors, including age, racial and ethnic diversity, and length of follow-up.
Those considering the implementation of BRCA1/2 screening programs should note two system factors that facilitated risk management performance and cancer diagnoses and undergird the genomic screening initiative: (i) an integrated health-care system capable of managing cases from genome-scale testing through evaluation and risk management and (ii) a learning health-care system committed to studying the outcomes of clinical interventions.20 A system that provides only surveillance and prophylactic surgery for individuals with genomic results cannot meaningfully contribute to evidence-based policy about the appropriateness of screening unselected individuals for genomic findings. In a learning health-care system, research and other learning activities are seamlessly integrated with standard and innovative care, all of which continuously inform one another. MyCode is a traditional genomic discovery research project, but actionable genomic results are returned to patient–participants who, supported by their clinicians, undergo traditional surveillance and prophylactic surgery. Effects on patient–participants and the health system of disclosing variants discovered via a genomic screening program (a form of innovative care) are studied (a learning activity) so that outcomes data can more readily inform clinical policies at our institution and beyond. Through this learning health-care system approach, we can learn whether returning BRCA1/2 findings to unselected individuals offers clinical utility.
Change history
13 October 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41436-021-01304-9
References
CDC. Public Health Genomics—Genomic Tests and Family History by Levels of Evidence.2014. https://www.cdc.gov/genomics/implementation/toolkit/tier1.htm.Accessed 28 April 2015.
King MC, Levy-Lahad E, Lahad A. Population-based screening for BRCA1 and BRCA2: 2014 Lasker Award. JAMA 2014;312:1091–1092.
Foulkes WD, Knoppers BM, Turnbull C. Population genetic testing for cancer susceptibility: founder mutations to genomes. Nat Rev Clin Oncol 2016;13:41–54.
Nelson HD, Pappas M, Zakher B, Mitchell JP, Okinaka-Hu L, Fu R. Risk assessment, genetic counseling, and genetic testing for BRCA-related cancer in women: a systematic review to update the U.S. Preventive Services Task Force recommendation. Ann Intern Med 2014;160:255–266.
Green RC, Berg JS, Grody WW et al, ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med 2013;15:565–574.
Carey DJ, Fetterolf SN, Davis FD et al, The Geisinger MyCode community health initiative: an electronic health record-linked biobank for precision medicine research. Genet Med 2016;18:906–913.
Dewey FE, Murray MF, Overton JD et al, Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science 2016;354.
Richards S, Aziz N, Bale S et al, Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424.
Murray MF. Your DNA is not your diagnosis: getting diagnoses right following secondary genomic findings. Genet Med 2016;18:765–767.
NCCN.Clinical Practice Guidelines in Oncology—Genetic/Familial High-Risk Assessment: Breast and Ovarian Version 2.2016. 2016. Accessed 21 October 2016.
Howlader N, Noone AM, Krapcho M et al, SEER Cancer Statistics Review, 1975-2013. 2016. http://seer.cancer.gov/csr/1975_2013/. Accessed 21 October 2016.
Narod SA, Iqbal J, Giannakeas V, Sopik V, Sun P. Breast cancer mortality after a diagnosis of ductal carcinoma in situ. JAMA Oncol 2015;1:888–896.
Finch AP, Lubinski J, Moller P et al, Impact of oophorectomy on cancer incidence and mortality in women with a BRCA1 or BRCA2 mutation. J Clin Oncol 2014;32:1547–1553.
Scheuer L, Kauff N, Robson M et al, Outcome of preventive surgery and screening for breast and ovarian cancer in BRCA mutation carriers. J Clin Oncol 2002;20:1260–1268.
Pinsky PF, Prorok PC, Kramer BS. Prostate cancer screening – a perspective on the current state of the evidence. N Engl J Med 2017;376:1285–1289.
Hosseini A, Khoury AL, Esserman LJ. Precision surgery and avoiding over-treatment. Eur J Surg Oncol 2017;43:938–943.
Buys SS, Partridge E, Black A et al, Effect of screening on ovarian cancer mortality: the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Randomized Controlled Trial. JAMA. 2011;305:2295–2303.
Sagara Y, Mallory MA, Wong S et al, Survival benefit of breast surgery for low-grade ductal carcinoma in situ: a population-based cohort study. JAMA Surg 2015;150:739–745.
Buchanan AH, Voils CI, Schildkraut JM et al, Adherence to recommended risk management among unaffected women with a BRCA mutation. J Genet Couns 2017;26:79–92.
IOM Genomics-Enabled Learning Health Care Systems: Gathering and Using Genomic Information to Improve Patient Care and Research: Workshop Summary. National Academies Press: Washington, DC, 2015.
Acknowledgements
The genomic screening program described here was funded by institutional support from GHS and grants from the Horace W. Goldsmith Foundation, the Mericle Foundation, the Robert Wood Johnson Foundation, and the Commonwealth of Pennsylvania—Department of Community and Economic Development. These funders had no role in study design, collection or analysis of data, or the decision to publish. We are grateful to the more than 150,000 MyCode participants for their participation in this research and, in particular, to the three patient–participants described here for their willingness to share their information. We thank the MyCode leadership, including Carroll Flansburg and Ann Mooney, and the many MyCode consenters, for their thorough patient engagement. We are grateful for the rigor and responsiveness exhibited by bioinformatics colleagues at the Laboratory for Molecular Medicine, particularly Christina Austin-Tse and Heidi Rehm, and at GHS, particularly Raghu Metpally and Thomas Person. Finally, we thank the staff of Geisinger Clinical Genomics and the Inherited Risk Breast Clinic for helping to coordinate care for individuals with a genomic result.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest as relates to the content in this article.
Additional information
Supplementary material is linked to the online version of the paper at
The original online version of this article was revised: The gene in panel B of Figure 1 was corrected to BRCA2
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Buchanan, A., Manickam, K., Meyer, M. et al. Early cancer diagnoses through BRCA1/2 screening of unselected adult biobank participants. Genet Med 20, 554–558 (2018). https://doi.org/10.1038/gim.2017.145
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2017.145
Keywords
This article is cited by
-
Returning individual genomic results to population-based cohort study participants with BRCA1/2 pathogenic variants
Breast Cancer (2023)
-
Observational study of population genomic screening for variants associated with endocrine tumor syndromes in a large, healthcare-based cohort
BMC Medicine (2022)
-
Addressing the routine failure to clinically identify monogenic cases of common disease
Genome Medicine (2022)
-
Implementing genomic screening in diverse populations
Genome Medicine (2021)
-
Pediatric reporting of genomic results study (PROGRESS): a mixed-methods, longitudinal, observational cohort study protocol to explore disclosure of actionable adult- and pediatric-onset genomic variants to minors and their parents
BMC Pediatrics (2020)
