
Abstract
Purpose:
Up to 1% of all men experience azoospermia, a condition of complete absence of sperm in the semen. The mechanisms and genes involved in spermatogenesis are mainly studied in model organisms, and their relevance to humans is unclear because human genetic studies are very scarce. Our objective was to uncover novel human mutations and genes causing azoospermia due to testicular meiotic maturation arrest.
Methods:
Affected and unaffected siblings from three families were subjected to whole-exome or whole-genome sequencing, followed by comprehensive bioinformatics analyses to identify mutations suspected to cause azoospermia. These likely mutations were further screened in azoospermic and normozoospermic men and in men proven to be fertile, as well as in a reference database of local populations.
Results:
We identified three novel likely causative mutations of azoospermia in three genes: MEIOB, TEX14, and DNAH6. These genes are associated with different meiotic processes: meiotic crossovers, daughter cell abscission, and possibly rapid prophase movements.
Conclusion:
The genes and pathways we identified are fundamental for delineating common causes of azoospermia originating in mutations affecting diverse meiotic processes and have great potential for accelerating approaches to diagnose, treat, and prevent infertility.
Genet Med advance online publication 16 February 2017
Similar content being viewed by others


Introduction
Approximately 15% of couples trying to conceive will not succeed after a year of attempts.1 Male factors account for approximately half of the cases.2 More than 50% of these cases remain idiopathic and 10–20% are azoospermic (complete absence of sperm in semen).3 Although this high frequency of human infertility has a notable heritable component,3,4 its genetic etiology remains unclear. Male infertility is usually due to a reduced amount of sperm cells (oligozoospermia) or their absence (azoospermia) in semen.
The most severe presentation of male infertility is nonobstructive azoospermia (NOA). Examination of testicular biopsy samples from NOA men reveals substantial histological variation commonly categorized into three groups: Sertoli cell–only (SCO), complete maturation arrest (MA), and mixed atrophy. The variation in the histopathology of the testis might be the consequence of the complexity of the spermatogenetic process and its variable genetic and epigenetic regulation.
Meiosis is a key gametogenesis process in which diploid germ cells undergo two reducing divisions to form haploid gametes (oocytes or sperm). In females, this process is initiated in the fetus, whereas meiosis in males is initiated at the onset of puberty and continues during their entire lifetime.5 In spermatogenesis, meiosis starts with several mitotic spermatogonial divisions with incomplete separation to form a syncytium of cells, producing primary spermatocytes that communicate with each other via cytoplasmic intercellular bridges.6 Primary spermatocytes then undergo the first meiotic division.
To ensure proper segregation of homologous chromosomes during the first meiotic division, these chromosomes physically pair and form chiasmata.7 A telomere-led rapid prophase chromosome movement is suggested to move them relative to one another in order to assist in establishing homologous interactions during pairing.8 The latter formation of chiasmata begins with the generation of programmed DNA double-strand breaks during the leptotene stage that are progressively repaired by homologous recombination during the zygotene and pachytene stages to complete the prophase stage.9,10
Each first meiotic division of spermatocytes yields a pair of secondary spermatocytes that then complete the second meiotic division. These haploid cells are called spermatids and they remain connected to each other through the cytoplasmic bridges, which allow them to function as diploid cells.11 Spermatids then mature into sperm cells that are released from the syncytium to the lumen of the seminiferous tubules. Errors in any of these meiotic processes may lead to spermatogenesis failure and to NOA. Even though the mechanisms and genes involved in these processes are well described in model organisms, their relevance to humans is poorly understood.
High frequency of aneuploidy, Y-chromosome microdeletions in the azoospermia regions,12 and reports of familial clustering of male infertility13,14,15 support the notion that genetic factors are major contributors to NOA. Copy-number variations, probably associated with idiopathic male infertility, have also recently been reported. (e.g., ref. 16). Variants in several other genes (e.g., MTHFR, FSHR, USP8, and TGFβ) have been found to be associated with increased risk for impaired spermatogenesis in reports that searched for variations on candidate genes in cohorts of infertile men or by genome-wide association studies (GWAS) (e.g., refs. 17,18,19). Putative mutations in TAF4B, ZMYND15, NPAS2, and TEX15 that most likely cause recessive familial impairments of spermatogenesis were recently detected by linkage analysis followed by next-generation exon sequencing.20,21,22
We and others have suggested that the relatively high incidence of NOA might stem from its being a group of complex pathologies of different genes, with at least a part of them being male-specific.4,17,23 However, there are currently only limited treatments and poor diagnostic tools for evaluating NOA patients. As a result, many of them undergo inevitably unnecessary testicular sperm extraction (TESE) surgeries.
To date, next-generation sequencing (NGS) technologies allow the derivation of the entire mutation landscape from desirable genomic segments. These advancements enhance the studies of Mendelian diseases.24 As a result, rapid identification of suspected causal mutations at single-nucleotide resolution became feasible, even in complex genetic backgrounds, thus avoiding the performance of linkage disequilibrium analysis of large family pedigrees.25
Here, we report a genetic study using NGS for three unrelated consanguineous families with familial NOA and testicular spermatocyte maturation arrest. We detected three novel variants in three genes not previously reported to be involved in men with NOA.
Materials and Methods
Patients and study population
The initial cohort included 1,000 infertile men referred to the institute for the study of fertility at the Tel Aviv Sourasky Medical Center during 1997–2010. The control group included 287 men with proven fertility, of whom 92 were sperm bank donors and 225 had at least two children. All infertile men underwent Y-chromosome microdeletion assessment and karyotype analyses. CFTR mutations, which pose a severe risk for obstructive azoospermia, were assessed according to clinical indications. All 92 sperm donors were normozoospermic (sperm concentration >40 × 106 per ml; sperm motility >50% during the first hour; normal morphology >4%26) and had normal karyotypes, no Y-chromosome microdeletion, and no familial history of infertility. All the men included in the study were queried about their parental origin, family ancestry, and fertility history. The ethnicity of the men was categorized as Arab or Jewish according to their parental origin. The former were Moslem, Christian, and Druze men, and the Jews were subdivided according to the country of ancestral origin. All the study participants consented to undergo genetic evaluation. The local institutional review board committee approved the study in accordance with the Helsinki Declaration of 1975.
Siblings from three families who reported familial infertility and had a similar histological testicular impairment were selected to undergo further genetic assessment by NGS.
Clinical features and biopsy evaluation
All analyzed siblings had azoospermia, except for one who had two sperm cells observed in one of the three sperm analyses that had been performed (Supplementary Table S1 online). The siblings had normal testicular volume and normal follicle-stimulating hormone (FSH) levels, except for one with slightly elevated FSH levels. None of them had Y-chromosome microdeletions.
After testicular sperm extraction (TESE) performed as previously reported,27 a small part of one biopsy sample was utilized for routine Bouin’s fixation for histopathological assessment and an additional part was minced and used to perform a meiosis evaluation by fluorescence in situ hybridization (FISH) as previously described.28,29 Briefly, testicular tissue was shredded and the released cells were washed and then resuspended and washed three times in 3:1 cold, fresh methanol acetic acid solution. The treated testicular cell suspension was stored at −20 °C until use. Prior to undergoing FISH, the treated cells were placed onto precleaned glass slides and air-dried. The slides were treated according to the manufacturer’s FISH instructions. Triple-color FISH with CEP centromeric DNA probes (Vysis, Downers Grove, IL) for chromosomes X, Y, and 18 (results for bivalent 18 are not included in the present report) and double-color FISH with TelVysion telomeric probes (Vysis) Xq/Yq (both orange) and Xp/Yp (both green) were used. Two independent observers viewed each slide with an Olympus AX70 fluorescence microscope (Olympus, Tokyo, Japan).
The nuclei of primary spermatocytes were identified by the typical granular or threadlike chromatin appearance of the DAPI counterstain. No selection was made for normal spermatocytes to additionally include moderately degenerated forms that characterize patients in the maturation arrest and mixed atrophy groups.
NGS data analysis
NGS input from whole-exome reads was aligned, a variants calling and genotyping file (VCF) was produced,30 and the variants were annotated23 and assessed for their relevance to the phenotype (see results for further description).
Conservation and selection analysis
Protein and nucleotide alignments of the candidate genes were retrieved from the UCSC genome browser, and sequences with large gaps were removed from the alignments. Selection analysis was performed using the Selecton server.31 Protein conservation and sequence domains were analyzed using the ConSurf,32 CDD,33 and InterPro34 servers.
Expression analysis
The RNAseq RPKM (reads per kilobase of transcript per million mapped reads) values were retrieved from the GTEx portal.35 A differential expression value was given to each gene as previously described.23 Briefly, a synthetic expression vector of exclusive expression in a tissue was created for all tested tissues. The Pearson product-moment correlation coefficient (r) was calculated for each synthetic vector against all the gene expression vectors. The results are a list of r-values indicating the expression specificity of each gene for each tissue.
Mutations analysis
DNA from all subjects was extracted from peripheral blood lymphocytes using the MasterPure Genomic DNA Purification Kit for blood (Epicenter, WI). Mutations suspected to cause azoospermia were confirmed by Sanger sequencing (Supplementary Table S2 online) and by examining the segregation within the respective families. Following this confirmatory step, the mutation frequency was assessed in ethnic populations selected from the cohort of 1,000 infertile men. Screening to detect each mutation in the population was performed in fertile and infertile men of the same ethnic origin to prevent erroneous findings by population stratification and in 22 additional infertile men who presented with complete spermatocyte maturation arrest. Mutations were detected by restriction analysis (RFLP) that was performed on PCR products and analyzed on agarose gel (2–4%, Supplementary Table S2 online). When the DNAH6 mutation was detected, an additional run on 10% acrylamide gel was performed to determine if the sample came from a heterozygote or a homozygote. Restriction sites for distinguishing between normal and mutant alleles were identified with the help of the following web servers: http://cedar.genetics.soton.ac.uk/public_html/primer.html, http://www.genscript.com/cgi-bin/tools/enzyme_cuttingtool, and http://www.justbio.com/cutter/index.php.
Results
Patient and control collection and characterization
We collected DNA samples from 1,000 Jewish and Arab men with azoospermia or severe oligozoospermia (<5 million sperm per ml) and from 227 matching fertile controls who fathered at least 2 children and 92 normozoospermic fertile men. Karyotype and carriers of CFTR mutations were reported in almost one-third of the infertile cohort. Familial infertility was reported in 10% of our patients, and parental consanguinity was present in 7.25% of them. We chose three families for which we had DNA samples for at least two phenotypically well-characterized siblings with a similar testicular impairment of complete spermatocyte maturation arrest (Supplementary Table S1 online). The rate of spermatocytes with XY chromosome bivalents was extremely low (in terms of both centromeres and telomeres being in proximity) in families A and C (Supplementary Table S3 online). Metaphase spermatocytes, identified by DAPI staining, were almost undetectable (2% of the spermatocytes in a sample of family member A.1 and none in the other three samples). XY chromosome bivalents were not assessed in family B, but its testicular histology revealed spermatogonia cells in all the tubules, with only a few spermatocytes in some of them. Groups of pyknotic nuclei, reminiscent of apoptosis or necrosis, were observed, particularly in some tubules in the family A specimens (Supplementary Figure S1 online). Sporadic cells were observed in family C samples, but none were observed in those from family B (Supplementary Table S1 online and Supplementary Figure S1 online). In addition, the collected cohorts served for screening the candidate mutations identified herein.
Overall, nine DNA samples were subjected to NGS. The seven subjects from families A and C underwent whole-exome sequencing, and the two siblings from family B underwent whole-genome sequencing.
Identification of mutations suspected to cause azoospermia
We set an analysis pipeline to identify new genes involved in familial infertility (Supplementary Figure S2 online). Whole-exome reads were aligned and a variants calling and genotyping file (VCF) was produced as described.30 Mutations corresponding to the inheritance model of each family and that do not appear in unaffected individuals were retrieved by a custom Perl script. All detected variants were comprehensively annotated, and the variation categories, transcript consequences, functional consequences, population frequencies, evolutionary conservation, and pathology information were added to each variant as previously reported.23 Likely causative mutations were identified by filtering the data according to (i) the recessive model of inheritance, (ii) the expected population frequency of the mutation (MAF <3%), and (iii) the functional impact of the mutation (conservation, loss of function, functional prediction). Finally, the relevance for the phenotype was assessed using comprehensive expression data,35 Gene Ontology terms,36 and model organism data.37
Mutations that passed these filtering steps were evaluated for their frequency in our in-house genome reference database that includes genotype–phenotype data for approximately 500 individuals from major Israeli ethnic groups (unpublished). The top suspected mutations then underwent validation by Sanger sequencing, familial segregation (when possible), and screening of patients with the same testicular impairment and fertile controls of the same ethnicity.
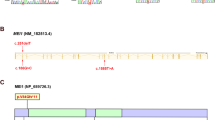
The whole-exome data from two affected brothers in family A included five mutations in five genes that passed all our filtration steps (Figure 2 and Supplementary Table S4 online). Segregation analysis of two additional affected brothers ruled out three of these mutations. The two remaining mutations are likely part of the same haplotype. The first mutation, which was in the gene pentraxin 4 (PTX4), was predicted to be damaging and had an approximately 0.5% population frequency in its corresponding ethnic group. Although pentraxin genes are associated with the immune response, PTX4 is found to have a relatively high expression in the testis as well as in the small intestine and brain.38 However, there is no evidence for the involvement of PTX4 in spermatogenesis. At 400 kb downstream to PTX4, we identified a novel, predicted deleterious asparagine-to-isoleucine nonsynonymous mutation in the MEIOB gene (NM_001163560:c.A191T:p.N64I). MEIOB seems exclusively expressed in adult human and mice testis (Figure 2a). In addition, we found asparagine 64 to be extremely conserved in metazoans (Figure 2b and Supplementary Figure S3 online). MeioB knockout mice39,40 are azoospermic due to meiotic arrest, similar to our patients ( Figure 1b ). These works suggested that MEIOB is required for double-strand break repair, crossover formation, and promotion of true and complete synapsis during meiosis. The identified mutation is located in one of three conserved DNA binding domains in MEIOB,39,40 and computational analysis suggests that this amino acid has structural importance ( Figure 2b ). The MEIOB mutation was further validated by Sanger sequencing ( Figure 1c ). RFLP and a computational screen of fertile and infertile males identified two additional patients as being homozygous to the mutation; they appeared to be the third and fourth affected siblings (matched by family and maiden name, Figure 1d ). No other carriers among our cohorts or in our reference database were identified (Supplementary Table S5 online).

Family A genetic and clinical findings. (a) Family A pedigree with black fill denoting azoospermic siblings. Brothers A.1 and A.2 underwent whole-exome sequencing. (b) Histologic section of patient A.1 showing spermatogenesis up to the spermatocyte cells (spc) stage at magnification ×400. (c) Sanger sequencing validation of the identified mutation in subject A.1. (d) RFLP analysis of all affected siblings.

Expression and conservation of the MEIOB gene. (a) MEIOB expression levels in men and women. RPKM values from 544 adult donors across 53 tissues retrieved from the GTEx project version 6. MEIOB is exclusively expressed in the testis. (b) MEIOB domain organization scheme and computational function analysis. RPA1 DBD is an OB fold ssDNA-binding domain of RPA1 proteins. The asterisk denotes the position of the mutated Asn 64. Results of ConSurf32 analysis of the Asn 64 region. Color intensity denotes conservation, “e” and “b” denote predicted exposed and buried residues, and ‘f’ and “s” denote predicted functional and structural important residues.
Whole-genome sequencing of two affected brothers from family B ( Figure 3 ) identified two mutations that passed all filtration steps. One mutation (NM_145292:exon5:c.G617C:p.G206A) was found in gene GALNTL5, which is expressed highly specifically in the testis. However, this mutation was found in a relatively high population frequency of 1.5% in the ethnic group of the affected family. In addition, another mutation in this gene was previously reported to cause impairment of sperm motility, but not to affect spermatogenesis. Thus, the relatively high population frequency of the mutation and the gene involvement in sperm motility, but not in early spermatogenesis, made this mutation less likely to be causative for NOA in this family. The second mutation we found was caused by a novel 10-bp deletion (NM_001201457:exon16: 2668-2678del) that led to a frameshift in gene TEX14, resulting in an early stop codon and a truncation of more than 500 codons before the wt termination codon. TEX14 was reported to be required for the formation of intercellular bridges in vertebrate germ cells, which are essential for meiosis during spermatogenesis.41 Severe spermatocyte depletion was observed in TEX14 KO mice and TEX14 exonic insertion in pigs.41,42 The porcine insertion occurs in exon 27, downstream to the deletion we identified here. It caused differential splicing of the exon and created a premature translation stop codon and an absence of protein product. TEX14 appears to be exclusively expressed in the testis of men and mice ( Figure 3e ) and is conserved among mammals. Further RFLP and computational screening of our cohorts and database did not find any other individuals who carried this mutation (Supplementary Table S5 online). Sanger sequencing validated this mutation ( Figure 3c ).

Family B genetic and clinical findings. (a) Family B pedigree with black fill denoting azoospermic siblings. Subjects B.1 and B.2 underwent whole-genome sequencing. (b) Histologic section of subject B.1 with spermatogonia (spg) in all the tubules and few spermatocyte cells (spc) at magnification ×400. (c) TEX14 domain architecture. “ANK rep” denotes an ankyrin repeat and “PKc like” denotes a protein kinase C-like domain. The asterisk marks the position of the identified deletion, and the resulting downstream truncated region is boxed and labeled. (d) Sanger sequencing validation of the identified mutation in subject B.1. (e) RFLP analysis of affected siblings. (f) TEX14 expression levels (shown as in Figure 2b). TEX14 is almost exclusively expressed in testis (light blue box) and, to some extent, in transformed fibroblasts of men and women (blue and red boxes, respectively), as labeled.
Family C was reported as a high parental consanguinity marriage ( Figure 4 ). We performed whole-exome sequencing for two infertile siblings, two healthy siblings, and one cousin with reported reduced sperm count who succeeded in fathering one child. Only one, rare, nonsynonymous substitution (MAF ~0.1%) in the gene DNAH6 passed all filtering steps (NM_001370:c.C10413A:p.H3471Q). This gene is overexpressed in human ( Figure 5a ) and mouse (http://biogps.org) testis. Extended segregation analysis of family C ( Figure 4d ) confirmed that only the affected azoospermic siblings were homozygous to the DNAH6 mutation. RFLP and computational screening of our cohorts and our reference database did not find any other homozygotes to this mutation (Supplementary Table S5 online). Homozygosity mapping43 found only one homozygous stretch shared in the affected brothers and absent in the unaffected family members ( Figure 5b ). This stretch covers our DNAH6 candidate mutation.

Family C genetic and clinical findings. (a) Family C pedigree with black fill denoting azoospermic members and grey fill denoting an oligozoospermic member. Subjects C.1 and C.2 were subjected to whole-exome sequencing. (b) Histologic section of patient C.1 showing spermatogenesis up to spermatocyte cells (spc) at magnification ×400. (c) Sanger sequencing validation of the identified mutation in subject C.1. (d) RFLP analysis of affected brothers C.1 and C.2 and other family members (C.3-C.8). Only the affected brothers were found to be homozygous for the suspected mutation, whereas all other members were carriers.

DNAH6 details. (a) DNAH6 is overexpressed in the testis. Red and blue bars denoting samples of women and men, respectively; pink and light blue boxes denote samples from reproductive tissues of women and men, respectively. (b) Genome-wide homozygosity mapping43 of family C reveals a significant block larger than 500 kb only in chromosome 2 (red bar). This block includes the DNAH6 locus. The X-axis denotes the physical position in each chromosome and the Y-axis denotes the homozygosity score, with the red and black bars indicating excess of and shortage of homozygosity blocks, respectively. (c) DNAH6 domain architecture and selection analysis. DHC N2, MT-binding, and D6 are dynein heavy chain N2, microtubule, and D6 domains, respectively, and the P-loop is an ATP phosphate-binding domain. Selecton31 analysis of human DNAH6 protein shows two clusters enriched in positively selected residues. The first cluster is very near the DNAH6 N-terminus and the second cluster includes the candidate mutation identified by us. The red dots mark positions of the identified positively selected residues. The grey inset details the positive selection in the region of the mutated H3471 residue colored according to the selection pressure.
DNAH6 is an axonemal dynein heavy chain suggested by homology as an inner dynein arm component.44 Dyneins are microtubule-associated motor protein complexes that generate force and movement on microtubules in various processes, including ciliary beating and cell division.45 DNAH6 knockdown disrupted motile cilia function in the human and mouse airway,46 but dynein genes have also been shown to be important for proper meiosis.47 In addition, no members of family C are known to have recurrent respiratory tract infections. DNAH6 protein and the candidate mutation amino acid position present an overall low conservation. Low conservation might stem from reduced functional importance or from nonadaptive processes, such as genetic drift, but it might also be driven due to adaptive evolution. To differentiate between these possibilities, we performed amino acid Ka/Ks tests, which compare the rates of synonymous (Ks) and nonsynonymous (Ka) substitutions and enable the inference of the selection direction (i.e., purifying, neutral or positive) and intensity.31 Positive selection (significance for Ka/Ks values >1) was found in 117 out of 4,158 (2.8%) amino acid positions, including the suspected mutation position ( Figure 5c ). More importantly, most of these 117 positively selected positions, including our suspected mutation position, were located in two dense clusters ( Figure 5c ). One cluster was located at the N terminus, and the other, which included the suspected mutation position, was located near the C terminus. This suggests that the relatively low conservation of the mutated amino acid is likely due to adaptation, and thus could be functionally important.
Conclusions
NOA can be caused by aneuploidy, Y-chromosome microdeletions,12 copy number variations,16 and mutations in specific genes.20,21,22 This supports genetic and heritable bases for NOA, but the origin for most cases remains elusive. Here, we studied NOA due to complete testicular meiotic maturation arrest in three families. We analyzed whole-exome or whole-genome sequencing and found novel likely NOA-causative mutations in the MEIOB, TEX14, and DNAH6 genes. None of these genes have ever been reported to cause NOA in humans. Each gene is associated with a different spermatogenesis pathway, demonstrating the complexity of azoospermia.
In family A ( Figures 1 and 2 ), we identified a novel, predicted deleterious (e.g., a change likely to alter the function of the protein, see Methods section), highly conserved asparagine-to-isoleucine mutation in the MEIOB gene. Meiob-deficient mice do not form crossovers in germ cells, and male and female Meiob knockout mice are infertile due to meiotic arrest.39,40 This is the phenotype observed in the affected brothers of family A. In adult humans, MEIOB seems to be exclusively expressed in the testis ( Figure 2a ). However, MEIOB was reported to be expressed in the fetal ovary, suggesting that it has a possible role in female meiosis.40 All four sisters of the affected brothers in family A are reported to have children. Because no DNA samples can be obtained from these sisters and other family members, we cannot assess the effect of the MEIOB mutation on women's fertility. Alignment of the X and Y chromosomes that occurs in the late zygotene or early pachytene stage48 probably precedes the γ-H2AX sex body staining stage.29 The low percentage of spermatocytes with X-Y chromosomes in proximity observed in brothers A1 and A2 is in agreement with the absence of sex body in pachytene-like spermatocytes in Meiob−/− mutant mice and with the suggestion that mice Meiob is required for chromosomal synapsis. We suggest that the NOA defect in family A is possibly due to problems in formation of meiotic crossovers or in paring of bivalents due to malfunction of the MEIOB protein in spermatocytes.
We found a novel deletion that leads to a frameshift and early stop codon in the TEX14 gene in family B ( Figure 3 ). An insertion in Tex14 exon 27, which resulted in a premature stop codon, was identified in several Finnish Yorkshire boars that had spermatogenic arrest in early meiotic cells.42 Spermatogenesis progressed to the early meiotic stage in TEX14 knockout mice, but it halted before the completion of the first meiotic division. In addition, TEX14 was required for formation and maintenance of the intercellular bridge and for cell abscission during meiosis in mice germ cells.41,49 Thus, in families A and B, we found comprehensive genetic and functional evidence pointing to our identified mutations as the causative ones for spermatogenic failure due to meiotic arrest present in our patients.
In family C, we detected a rare nonsynonymous mutation in the poorly characterized heavy axonemal dynein chain gene DNAH6. Homozygosity mapping followed by extended segregation analysis in eight family members confirmed this finding and identified a genomic segment covering DNAH6 as the only region to fit the recessive model of inheritance in this family. DNAH6 is only known to be a member of the dynein proteins family;50 there is no information regarding its function in general or specifically in spermatogenesis. A possible hint comes from a recent study that found co-occurrence of a large deletion in the human DNAH6 locus and primary ovarian insufficiency.51 The high rate of amino acid substitutions during mammalian evolution of the DNAH6 mutated amino acid region might be mainly due to adaptive selection, a feature highly associated with some genes involved in reproduction.52,53,54 Notably, dynein genes have been suggested as the main components of the force generating rapid prophase movements of the chromosomes (RPMs) during meiosis in various phylogenetic classes.8,55 RPMs have been proposed to move chromosomes relative to one another, helping proper homologous pairing, resolving chromosome entanglements, and regulating chiasma placements.8 However, as far as we know, the exact dynein genes involved in mammalian RPMs have not been identified. In this context, the function of DNAH6 should be further investigated.
In conclusion, we found different likely NOA-causative mutations in different genes that associate with different pathways of the first meiosis in each of the studied families. This highlights the effectiveness and value of advanced genetic and genomic analyses of familial male infertility cases and their usefulness in improving diagnoses of and treatment procedures for this condition and in important basic science discoveries.
Disclosure
The authors declare no conflict of interest.
References
de Kretser DM. Male infertility. Lancet 1997;349:787–790.
Farhi J, Ben-Haroush A. Distribution of causes of infertility in patients attending primary fertility clinics in Israel. Isr Med Assoc J 2011;13:51–54.
Byler MC, Lebel RR. Risks of reproducing with a genetic disorder. Semin Reprod Med 2013;31:258–266.
Hwang K, Yatsenko AN, Jorgez CJ, et al. Mendelian genetics of male infertility. Ann N Y Acad Sci 2010;1214:E1–E17.
Feng CW, Bowles J, Koopman P. Control of mammalian germ cell entry into meiosis. Mol Cell Endocrinol 2014;382:488–497.
Gilbert, S. Developmental Biology. Sinauer Associates: Sunderland, MA, 2000.
Bascom-Slack CA, Ross LO, Dawson DS. Chiasmata, crossovers, and meiotic chromosome segregation. Adv Genet 1997;35:253–284.
Koszul R, Kleckner N. Dynamic chromosome movements during meiosis: a way to eliminate unwanted connections? Trends Cell Biol 2009;19:716–724.
Kauppi L, Jasin M, Keeney S. How much is enough? Control of DNA double-strand break numbers in mouse meiosis. Cell Cycle 2013;12:2719–2720.
Weiner BM, Kleckner N. Chromosome pairing via multiple interstitial interactions before and during meiosis in yeast. Cell 1994;77:977–991.
Braun RE, Behringer RR, Peschon JJ, Brinster RL, Palmiter RD. Genetically haploid spermatids are phenotypically diploid. Nature 1989;337:373–376.
O’Flynn O’Brien KL, Varghese AC, Agarwal A. The genetic causes of male factor infertility: a review. Fertil Steril 2010;93:1–12.
Gianotten J, Westerveld GH, Leschot NJ, et al. Familial clustering of impaired spermatogenesis: no evidence for a common genetic inheritance pattern. Hum Reprod 2004;19:71–76.
van Golde RJ, van der Avoort IA, Tuerlings JH, et al. Phenotypic characteristics of male subfertility and its familial occurrence. J Androl 2004;25:819–823.
Inhorn MC, Kobeissi L, Nassar Z, Lakkis D, Fakih MH. Consanguinity and family clustering of male factor infertility in Lebanon. Fertil Steril 2009;91:1104–1109.
Eggers S, DeBoer KD, van den Bergen J, et al. Copy number variation associated with meiotic arrest in idiopathic male infertility. Fertil Steril 2015;103:214–219.
Kosova G, Scott NM, Niederberger C, Prins GS, Ober C. Genome-wide association study identifies candidate genes for male fertility traits in humans. Am J Hum Genet 2012;90:950–961.
Tapanainen JS, Aittomäki K, Min J, Vaskivuo T, Huhtaniemi IT. Men homozygous for an inactivating mutation of the follicle-stimulating hormone (FSH) receptor gene present variable suppression of spermatogenesis and fertility. Nat Genet 1997;15:205–206.
Wu W, Shen O, Qin Y, et al. Methylenetetrahydrofolate reductase C677T polymorphism and the risk of male infertility: a meta-analysis. Int J Androl 2012;35:18–24.
Ayhan Ö, Balkan M, Guven A, et al. Truncating mutations in TAF4B and ZMYND15 causing recessive azoospermia. J Med Genet 2014;51:239–244.
Okutman O, Muller J, Baert Y, et al. Exome sequencing reveals a nonsense mutation in TEX15 causing spermatogenic failure in a Turkish family. Hum Mol Genet 2015;24:5581–5588.
Ramasamy R, Bakırcıoğlu ME, Cengiz C, et al. Whole-exome sequencing identifies novel homozygous mutation in NPAS2 in family with nonobstructive azoospermia. Fertil Steril 2015;104:286–291.
Gershoni M, Pietrokovski S. Reduced selection and accumulation of deleterious mutations in genes exclusively expressed in men. Nat Commun 2014;5:4438.
Ku CS, Naidoo N, Pawitan Y. Revisiting Mendelian disorders through exome sequencing. Hum Genet 2011;129:351–370.
Schneeberger K. Using next-generation sequencing to isolate mutant genes from forward genetic screens. Nat Rev Genet 2014;15:662–676.
World Health Organization. WHO Laboratory Manual for the Examination and Processing of Human Semen. 2010. http://www.who.int/reproductivehealth/publications/infertility/9789241547789/en/.
Hauser R, Botchan A, Amit A, et al. Multiple testicular sampling in non-obstructive azoospermia–is it necessary? Hum Reprod 1998;13:3081–3085.
Yogev L, Gamzu R, Paz G, et al. Rate of homologous chromosome bivalents in spermatocytes may predict completion of spermatogenesis in azoospermic men. Hum Genet 2002;110:30–35.
Yogev L, Zeharia E, Kleiman SE, et al. Use of sex chromosome bivalent pairing in spermatocytes of nonobstructive azoospermic men for the prediction of successful sperm retrieval. Fertil Steril 2006;86:106–112.
Oz-Levi D, Ben-Zeev B, Ruzzo EK, et al. Mutation in TECPR2 reveals a role for autophagy in hereditary spastic paraparesis. Am J Hum Genet 2012;91:1065–1072.
Stern A, Doron-Faigenboim A, Erez E, Martz E, Bacharach E, Pupko T. Selecton 2007: advanced models for detecting positive and purifying selection using a Bayesian inference approach. Nucleic Acids Res 2007;35(Web Server issue):W506–W511.
Ashkenazy H, Abadi S, Martz E, et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 2016;44(W1):W344–W350.
Marchler-Bauer A, Derbyshire MK, Gonzales NR, et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res 2015;43(Database issue):D222–D226.
Mitchell, A, Chang HY, Daugherty L, et al. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res 2015; 43(Database issue):D213–D221.
Ardlie, KG, Deluca DS, Segrè AV, et al.; GTEx Consortium. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015;348:648–660.
Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000;25:25–29.
Eppig JT, Blake JA, Bult CJ, Kadin JA, Richardson JE ; Mouse Genome Database Group. The Mouse Genome Database (MGD): facilitating mouse as a model for human biology and disease. Nucleic Acids Res 2015;43(Database issue):D726–D736.
Martinez de la Torre Y, Fabbri M, Jaillon S, et al. Evolution of the pentraxin family: the new entry PTX4. J Immunol 2010;184:5055–5064.
Luo M, Yang F, Leu NA, et al. MEIOB exhibits single-stranded DNA-binding and exonuclease activities and is essential for meiotic recombination. Nat Commun 2013;4:2788.
Souquet B, Abby E, Hervé R, et al. MEIOB targets single-strand DNA and is necessary for meiotic recombination. PLoS Genet 2013;9:e1003784.
Greenbaum MP, Yan W, Wu MH, et al. TEX14 is essential for intercellular bridges and fertility in male mice. Proc Natl Acad Sci USA 2006;103:4982–4987.
Sironen A, Uimari P, Venhoranta H, Andersson M, Vilkki J. An exonic insertion within Tex14 gene causes spermatogenic arrest in pigs. BMC Genomics 2011;12:591.
Seelow D, Schuelke M, Hildebrandt F, Nürnberg P. HomozygosityMapper–an interactive approach to homozygosity mapping. Nucleic Acids Res 2009;37(Web Server issue):W593–W599.
Hom EF, Witman GB, Harris EH, et al. A unified taxonomy for ciliary dyneins. Cytoskeleton (Hoboken) 2011;68:555–565.
Roberts AJ, Kon T, Knight PJ, Sutoh K, Burgess SA. Functions and mechanics of dynein motor proteins. Nat Rev Mol Cell Biol 2013;14:713–726.
Li, Y. Yagi H, Onuoha EO, et al. DNAH6 and its interactions with PCD genes in heterotaxy and primary ciliary dyskinesia. PLoS Genet 2016;12:e1005821.
McNally FJ. Mechanisms of spindle positioning. J Cell Biol 2013;200:131–140.
Solari AJ. Synaptosomal complexes and associated structures in microspread human spermatocytes. Chromosoma 1980;81:315–337.
Iwamori T, Iwamori N, Ma L, Edson MA, Greenbaum MP, Matzuk MM. TEX14 interacts with CEP55 to block cell abscission. Mol Cell Biol 2010;30:2280–2292.
Vaughan KT, Mikami A, Paschal BM, et al. Multiple mouse chromosomal loci for dynein-based motility. Genomics 1996;36:29–38.
Norling A, Hirschberg AL, Rodriguez-Wallberg KA, Iwarsson E, Wedell A, Barbaro M. Identification of a duplication within the GDF9 gene and novel candidate genes for primary ovarian insufficiency (POI) by a customized high-resolution array comparative genomic hybridization platform. Hum Reprod 2014;29:1818–1827.
Jansa SA, Lundrigan BL, Tucker PK. Tests for positive selection on immune and reproductive genes in closely related species of the murine genus mus. J Mol Evol 2003;56:294–307.
Wagner A. Rapid detection of positive selection in genes and genomes through variation clusters. Genetics 2007;176:2451–2463.
Wyckoff GJ, Wang W, Wu CI. Rapid evolution of male reproductive genes in the descent of man. Nature 2000;403:304–309.
Lee CY, Horn HF, Stewart CL, et al. Mechanism and regulation of rapid telomere prophase movements in mouse meiotic chromosomes. Cell Rep 2015;11:551–563.
Acknowledgements
This work was funded by the Weizmann Institute of Science Kekst Family Center for Medical Genetics, the David and Fela Shapell Family Center for Genetic Disorders Research, and the Crown Human Genome Center.
Author information
Authors and Affiliations
Corresponding authors
Supplementary information
Supplementary Figures and Tables
(DOCX 2974 kb)
Rights and permissions
About this article

Cite this article
Gershoni, M., Hauser, R., Yogev, L. et al. A familial study of azoospermic men identifies three novel causative mutations in three new human azoospermia genes. Genet Med 19, 998–1006 (2017). https://doi.org/10.1038/gim.2016.225
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2016.225
Keywords
This article is cited by
-
Elucidation of population stratifying markers and selective sweeps in crossbred Landlly pig population using genome-wide SNP data
Mammalian Genome (2024)
-
Sertoli cell-only syndrome: advances, challenges, and perspectives in genetics and mechanisms
Cellular and Molecular Life Sciences (2023)
-
Novel copy number variations within SYCE1 caused meiotic arrest and non-obstructive azoospermia
BMC Medical Genomics (2022)
-
Identification of deleterious variants in patients with male infertility due to idiopathic non-obstructive azoospermia
Reproductive Biology and Endocrinology (2022)
-
Diagnostica genetica dell’infertilità maschile: nuovi approcci
L'Endocrinologo (2022)
