
Abstract
Background:
Genetic FBN1 testing is pivotal for confirming the clinical diagnosis of Marfan syndrome. In an effort to evaluate variant causality, FBN1 databases are often used. We evaluated the current databases regarding FBN1 variants and validated associated phenotype records with a new Marfan syndrome geno-phenotyping tool called the Marfan score.
Methods and results:
We evaluated four databases (UMD-FBN1, ClinVar, the Human Gene Mutation Database (HGMD), and Uniprot) containing 2,250 FBN1 variants supported by 4,904 records presented in 307 references. The Marfan score calculated for phenotype data from the records quantified variant associations with Marfan syndrome phenotype. We calculated a Marfan score for 1,283 variants, of which we confirmed the database diagnosis of Marfan syndrome in 77.1%. This represented only 35.8% of the total registered variants; 18.5–33.3% (UMD-FBN1 versus HGMD) of variants associated with Marfan syndrome in the databases could not be confirmed by the recorded phenotype.
Conclusion:
FBN1 databases can be imprecise and incomplete. Data should be used with caution when evaluating FBN1 variants. At present, the UMD-FBN1 database seems to be the biggest and best curated; therefore, it is the most comprehensive database. However, the need for better genotype–phenotype curated databases is evident, and we hereby present such a database.
Genet Med advance online publication 01 December 2016
Similar content being viewed by others


Introduction
The first systematic definition of Marfan syndrome (MFS) was the so-called Berlin criteria1 of 1986. After the discovery of FBN1 as the disease-causing gene for MFS in 1991,2 the criteria were revised in Ghent in 1996 (Ghent I).3 In 2010, the criteria were again revised (Ghent II), highlighting FBN1 mutations, aortic dilatation, and ectopia lentis as cornerstones in the MFS diagnosis.4
According to the Ghent II criteria, it is possible to diagnose MFS by evaluating clinical manifestations, but genetic testing for diagnosing MFS has proven to be increasingly important.5 According to the American College of Medical Genetics and Genomics (ACMG),6 a variant should be classified with one of the following modifiers: (i) pathogenic, (ii) likely pathogenic, (iii) uncertain significance, (iv) likely benign, or (v) benign. ACMG suggests that the term “likely” be used when a variant is at least 90% likely to be either benign or pathogenic and, of course, with even stronger evidence for the terms “benign” or “pathogenic.” Variants with insufficient evidence are termed variants of uncertain significance (VUS). In clinical practice, a variant evaluation that results in a VUS statement is of minor diagnostic utility.7 When categorizing a variant, a VUS classification calls for gathering sufficient additional evidence in the direction of either “benign” or “pathogenic.”.
The Ghent II criteria for causality of variants4 in the FBN1 gene are listed in a box in the original guideline paper and repeated in the general guideline text. Some of the them, including “nonsense mutations,” “in-frame and out-of-frame deletion/insertion,” and “mutations previously shown to segregate in the Marfan family” are generally accepted in the genetics community.6,8 Others are specific for the FBN1 gene, such as “missense affecting/creating cysteine residues”4 and “missense affecting conserved residues of the EGF consensus sequence ((D/N)X(D/N)(E/Q)Xm(D/N)Xn(Y/F), with m and n representing variable numbers of residues: D, aspartic acid; N, asparagine; E, glutamic acid; Q, glutamine; Y, tyrosine; F, phenylalanine).”4 No references to substantiate these recommendations are cited, but some of the recommendations (for example, regarding the effect of cysteine mutations) were inspired by the work by Faivre et al.,9 which was published a few years before the guidelines. According to the OMIM database (http://omim.org), the FBN1 gene is associated with no fewer than nine conditions (acromicric dysplasia, familial thoracic aortic aneurysm, familial ectopia lentis, geleophysic dysplasia 2, MFS, MASS phenotype, Scheuermann kyphosis, stiff skin syndrome, and Weill–Marchesani syndrome 2), some of which are in no way like MFS. Thus, the presence of a causal variant in the FBN1 gene is not necessarily associated with or causes MFS. In reality, laboratories seldom report results referring to the Ghent II variant criteria, and it seems that each laboratory has its own criteria for evaluating variant causality, and even the nomenclature can be troublesome for some laboratories.10
The variant evaluation process is usually performed according to local and generalized guidelines used to evaluate any kind of variant. Variant databases such as the Human Gene Mutation Database (HGMD) are used as a resource for linkage to peer-reviewed articles on disease-associated variants,11 but these databases have shown inaccuracies when used to diagnose MFS.12 Sequencing techniques such as next-generation sequencing and the use of large gene panels as well as whole-exome sequencing increase the demand for complex computer algorithms to handle data from various sources, including genotype–phenotype databases.11 Therefore, a well-curated variant database focused on genotype–phenotype correlation is essential when evaluating previously described variants.
We hereby provide a comprehensive quality evaluation of the present databases with information on FBN1 variants as well as variant-associated phenotypes and introduce a new MFS genotype–phenotype tool called the Marfan score.
Materials and Methods
We created a database with all publicly available FBN1 variants and all known associated case-based MFS phenotype data. All data were manually evaluated on a case-based level, and relevant phenotype data according to the Ghent II nosology4 were extracted and entered into our new database. Each record was linked to a reference representing the source of the record. We searched the Universal Mutation Database for FBN1 (UMD-FBN1; http://www.umd.be/FBN1/),13 the HGMD professional database (http://www.hgmd.cf.ac.uk/ac/index.php), the ClinVar database (National Center for Biotechnology Information; http://www.ncbi.nlm.nih.gov/clinvar/),14 and the Universal Protein Resource (UniProt; http://www.uniprot.org/) database15 for all known FBN1 variants. In each database, we identified reference articles and other material in as much detail as possible. Published peer-reviewed articles were all identified by PubMed searches. All material written in English was evaluated; nine papers in Chinese were not evaluated. It was not possible to gain access to 12 papers indexed in PubMed, representing 15 of 4,904 entries in the database.
The UMD-FBN1 mutations database also contains data classified as “personal communication,” which is not published in the literature and is therefore accessible only at the database’s homepage (http://www.umd.be/FBN1/).
Evaluating papers for variants referred in the databases, we found additional variants (n = 168) that, for unknown reasons, were not registered in any of the databases. These variants were as well registered in our database and evaluated in the current setup.
Each record for a variant was regarded as a specific individual representing a specific phenotype. We classified records only when it was possible to identify a specified individual representing a specific phenotype. In cases of multiple reports for the same individual, only one record was evaluated. For all individuals reported more than once in the literature, the report with the most detailed phenotype was used and no other record representing the same individual was evaluated.
Each record was classified into one of seven groups:
-
1. “Nonclassified,” representing records in which no phenotypic data were available or the recorded individual was already registered in the database
-
2. “Polymorphism,” representing records stating that the variant was found in individuals not having MFS or stated it as a polymorphism
-
3. “MFS Berlin,” representing records without detailed phenotypical data but describing the individual as fulfilling the Berlin criteria of MFS1
-
4. “MFS Ghent I,” representing records without detailed phenotypical data but describing the individual as fulfilling the first revised Ghent criteria of MFS3
-
5. “MFS Ghent II,” representing records without detailed phenotypical data but describing the individual as fulfilling the second revised Ghent criteria of MFS4
-
6. “Incomplete MFS,” representing records without detailed phenotypical data but describing the individual as having incomplete MFS, with MFS habitus, MFS-like phenotype, or something else
-
7. “Clinical classification,” representing records with phenotypic data
During evaluation of FBN1 databases, we registered whether the database associated the variant with MFS. If the variant was associated with MFS at least once, then we defined the variant as a database MFS diagnosis (database-MFS).
Marfan score
To provide phenotypical data with a numeric value when handling multiple references, the Marfan score was established. A numeric score was chosen because the databases often have numerous records for the same variant when the phenotype information points toward differing effects in different patients and because phenotype information for individual patients is often incomplete. A numeric Marfan score enables the management of such scenarios because it can handle variant associations with MFS and specific and relevant phenotypes, and it differentiates references with good phenotype descriptions from those with unspecific and insufficient descriptions. The present Marfan score is not intended to be a tool used in daily clinical practice. At this time, the Marfan score should be used only to evaluate the feasibility and quality of current databases that include information on MFS.
For all cases classified as “clinical classification,” the Marfan score ( Table 1 ) was based on the “systemic criteria” in the Ghent II nosology,4 which is based on the provided clinical data. Because aortic dilatation/dissection is not among the systemic criteria in the Ghent II nosology but is still a very important clinical feature, we chose to score aorta dilation/dissection as 10 points. Moreover, a causal FBN1 variant combined with aortic dilatation are sufficient for diagnosing MFS according to Ghent II nosology.4
We also considered the clinical presentation of a patient with a clear MFS phenotype as the best marker for the genotype–phenotype association of a variant and MFS. To highlight the effect of high-quality phenotype records and to ensure that these records were reflected in the mean of a variant’s Marfan score, cases fulfilling the Ghent II criteria—based solely on phenotypical data (e.g., lens luxation and aorta dilatation)—were scored an additional 20 points.
We considered a Marfan score ≥7 points as indicating an association with MFS (score-MFS), but it is obvious that a higher Marfan score had greater association significance than a lower score. In theory, it is possible to have a Marfan score >7 points without fulfilling the Ghent II criteria, but a score of 7 points would be found only in patients with at least some phenotypical characteristics typical for MFS. Therefore, a cutoff of 7 points is a conservative estimate when evaluating database diagnoses.
For all cases defined as a “polymorphism,” “MFS Berlin,” “MFS Ghent I,” “MFS Ghent II,” or “incomplete MFS,” the Marfan score was defined as shown in Table 1 . No nonclassified records were scored.
Results
Database
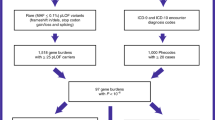
We registered 2,250 FBN1 variants in our data set based on 4,904 single records presented in 307 references. It was possible to identify a specific individual in 2,303 records. For the remaining 2,601 records, either we were not able to determine whether the records represented a specific individual or we identified the records as representing an already published individual. We found 168 variants in searched references that were not registered in the databases, resulting in 2,082 database variants.
Database-MFS and score-MFS
Of the total 2,082 variants ( Figure 1 and Table 2 , row 1), we were able to calculate a Marfan score for 1,283 variants, of which 69.7% (n = 893) had a Marfan score ≥7 and were therefore regarded as score-MFS. These were distributed as 830 (74.6% of all variants possible for the Marfan score in the database) in UMD-FBN1, 542 (73.3%) in HGMD, 73 (57.9%) in ClinVar, and 171 (69.7%) in Uniprot.

Percentages of all variants in different subgroups. “Database-Marfan syndrome” (MFS) represents variants associated with MFS in at least one FBN1 database. “Possible to Marfan score” represents all variants classifiable with a Marfan score. “Score-MFS” represents variants with a Marfan score ≥7. “Database-MFS” and “score-MFS” represent all variants associated with MFS in at least one FBN1 database and with a Marfan score ≥7.
In all four databases, 1,661 variants ( Figure 1 and Table 2 , row 3) were registered at least once as being associated with MFS; therefore, they were regarded as database-MFS. In general, the Marfan scores were higher for database-MFS, with mean Marfan scores ranging from 10.51 in ClinVar to 13.64 in UMD-FBN1, compared with non-database MFS variants with mean Marfan scores ranging from −7.16 in ClinVar to 8.67 in UMB-FBN1. The negative ClinVar figure indicates that the database contains a high percentage of variants correctly not linked to MFS.
When evaluating database-MFS versus score-MFS correlations, we could only confirm score-MFS in 746 database-MFS variants (35.8% of all variants ( Figure 1 ), representing 77.1% of all variants for which assignment of a Marfan score was possible. In the specific databases, there were 612 (81.5% of all classifiable database-MFS variants in the database) in UMD-FBN1, 516 (76.7 %) in HGMD, 71 (68.3%) in ClinVar, and 169 (76.8%) in Uniprot ( Table 2 (row 8)). Most of the variants appear in several of the databases.
If we accepted a Marfan score cutoff of ≤7 as a marker for no phenotypical association of the variant with MFS (non-score-MFS), then the UMD-FBN1 database associated variants with MFS without clinical evidence for 18.5% of variants, HGMD did so for 33.3%, ClinVar for 31.7%, and UniProt for 23.2% ( Table 2 , row 9). When evaluating score-MFS with non-database-MFS, we found score-MFS for 54.4% of all scoreable non-database-MFS. This indicates that databases contain incorrect conclusions for variants.
Supplementary data
Individual variants and the data used for calculating the Marfan score are provided in Supplementary Data S1 online. We searched the ExAC (Exome Aggregation Consortium) data set16for allele frequency data for each recorded variant. Supplementary Data S2 presents these data. We manually searched all recorded variants for the in silico scores of SIFT,17 Mutation Taster,18 and PhyloP using the Alamut v2.3 software package (Interactive Biosoftware, Rouen, France). We also manually searched all nonsynonymous variants for PolyPhen 2 HumDiv via the PolyPhen 2 homepage. Supplementary Data S3 online contains the in silico score data. When available in the databases, we also recorded expected variant effects on amino acids. Supplementary Data S4 online shows the collected amino acid data.
Discussion
At present, the four databases collectively provide the diagnostic tools for evaluating genetic test results when diagnosing MFS. Based on these four databases, we compiled a large, comprehensive, and well-curated FBN1 database with detailed descriptions of genotype–phenotype relations. We also defined a new Marfan score to test currently used databases to gauge the quality of the information in these curated databases. MFS is a well-defined monogenic disorder with a widely accepted systematic phenotypical scoring criteria described in the Ghent II nosology.4 The Marfan score is an effort to operationalize the MFS phenotype in one figure in accordance with the diagnostic criteria of Ghent II. To minimize loss of valuable data in the Marfan score, we also used imprecise information such as “incomplete Marfan” or “classical Marfan.” We regarded the Ghent II criteria as the gold standard for diagnosing MFS, but older criteria or imprecise definitions also affect the Marfan score.
Our evaluation of the four FBN1 variant databases showed that they have different characteristics. We considered the size of the database as a key parameter because the likelihood that a variant was in a database must be correlated with the size of the database. However, the number of unique variants could also be important because the variant undergoing evaluation could be a unique variant represented in only one database.
We observed large differences between databases regarding the absolute number as well as the percentage of unique variants. The size of the database was not an indicator of the proportion of unique variants because HGMD had only 5.0% of the total unique variants even though the database contained 47.5% of the total variants. Strikingly, 60.2% of all variants in the rather small ClinVar database were unique variants. Still, many unique variants do not necessarily indicate that the database is better than other databases because variants without supporting data are of very little value. Our data indicate that the ClinVar database did indeed have many unique variants, but a majority (61.7%) of variants could not be classified with a Marfan score based on the available data.
Only 47 variants were present in all four databases. It is therefore very important to use more databases when looking for variant data. We cannot recommend using any of the databases as a sole indicator of variant pathogenicity. However, the current collated database could prove to be an important tool for diagnosing MFS in the future.
We found 168 variants in searched references that were not recorded in any of the databases. None of the variants was clearly associated with MFS, and we could score only 63 variants, of which 61 scored as polymorphisms, 1 variant scored 1 point due to mitral prolapse, and 1 variant scored 2 points due a registration of “incomplete MFS.” This shows that relevant published data overall are recorded in the databases.
The databases provided very little information regarding the variants registered in the database. Only 61.6% of the registered variants were classifiable with a Marfan score, indicating that the databases have many variants registered without accessible documentation for any disease-causing effect. There was a large variation in classifiable variants between the databases, with Uniprot having 91.3% of the registered variants classifiable with a Marfan score compared with only 38.3% in ClinVar. Both databases have a low number of registered variants compared with UMD-FBN1 and HGMD, which could explain the magnitude of variance. The fact that the smallest database (Uniprot) also had the highest degree of phenotypes that we were able to score could be explained by the fact that large databases may be less critical with the data they record.
We have found that laboratories, when provided with genetic material to analyze, do not receive comprehensive phenotype data from the referring clinicians. For this reason, they cannot provide detailed variant-associated phenotype data to the databases. This is a major reason why databases contain large quantities of variant data but not of appropriate and detailed phenotype data.
In evaluating database-MFS, it is alarming that up to one-third of variants (in the HGMD) do not score enough points with the conservative cutoff limit of ≥7 points in score-MFS. The database-MFS and score-MFS correlation proportion represent only 35.8% of all registered variants ( Figure 1 ), indicating that an even larger proportion of the information in the databases is based on undocumented diagnosis statements than we can evaluate using the Marfan score.
As more genetic tests are being performed owing to reduced costs and easier access to sequencing facilities, we expect that new variants will be discovered more frequently. We also predict a more frequent single-patient setting where segregation data are unavailable. We therefore expect an increasing demand for fast and reliable classification of variants in diagnostic laboratories. It is our impression that variant databases, at least to some extent, are already used by laboratories when analyzing variants. To our knowledge, there are no data regarding how precise these databases are when used as a diagnostic tool, but the present data indicate that one should be cautious when using these databases.
Evaluation of reference literature regarding MFS and associated variants is an expert effort because the genotype–phenotype association for specific variants must be conducted by an evaluator with specific knowledge of the MFS phenotype as well as the history of the diagnostic criteria. Clinical manifestations may also vary considerably, both interfamiliarly and intrafamiliarly,19 making it even more difficult for non-MFS experts to determine whether a genotype–phenotype exists.
ACMG accepts the use of databases as supporting evidence but warns about “how frequently the database is updated, whether data curation is supported, and what methods were used for curation.”6 Previously, we showed that all current databases include data of equivocal quality.12 The arbitrary goal of 90% certainty that a variant is either likely benign or likely pathogenic is often difficult or impossible to achieve for missense variants when following the ACMG 2015 guidelines. Still, the ACMG guidelines recommend that laboratories use and report to variant databases, including the ClinVar database.6 In the present study, we did not find any data that could specifically support use of the ClinVar database for assessing FBN1 variants. Instead, we recommend well-curated databases based on phenotype data associated with each variant. We also suggest using some sort of phenotype scoring system and propose our Marfan score as a method for scoring FBN1 variants associated to MFS. The database data used in this study can be found in the Supplementary Information and are free to use as a transient update and curation of the FBN1 databases. We also provide three separate supplementary variant data sets: one for ExAC allele frequency;16 one for expected amino acid effects; and one for in silico scores from SIFT,17 Mutation Taster,18 PhyloP, and PolyPhen 2 HumDiv.20,21
Conclusion
The current FBN1 databases should be used with caution when evaluating FBN1 variant pathogenicity. Therefore, it is of great importance to use more than one database when searching for variant data. At present, the UMD-FBN1 database seems to be the biggest and best curated and therefore the most comprehensive database. However, the need is evident for better-curated databases containing clear phenotype–genotype associations. A systematic phenotype scoring system could aid in clinical decision making.
Disclosure
The authors declare no conflict of interest.
References
Beighton P, de Paepe A, Danks D, et al. International Nosology of Heritable Disorders of Connective Tissue, Berlin, 1986. Am J Med Genet 1988;29:581–594.
Dietz HC, Cutting GR, Pyeritz RE, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 1991;352:337–339.
De Paepe A, Devereux RB, Dietz HC, Hennekam RC, Pyeritz RE. Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet 1996;62:417–426.
Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet 2010;47:476–485.
Sheikhzadeh S, Kade C, Keyser B, et al. Analysis of phenotype and genotype information for the diagnosis of Marfan syndrome. Clin Genet 2012;82:240–247.
Richards S, Aziz N, Bale S, et al.; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424.
von Kodolitsch Y, Kutsche K. Interpretation of sequence variants of the FBN1 gene: analog or digital? A commentary on decreased frequency of FBN1 missense variants in Ghent criteria-positive Marfan syndrome and characterization of novel FBN1 variants. J Hum Genet 2015;60:465–466.
Plon SE, Eccles DM, Easton D, et al.; IARC Unclassified Genetic Variants Working Group. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat 2008;29:1282–1291.
Faivre L, Collod-Beroud G, Loeys BL, et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet 2007;81:454–466.
Deans ZC, Fairley JA, den Dunnen JT, Clark C. HGVS Nomenclature in practice: an example from the United Kingdom National External Quality Assessment Scheme. Hum Mutat 2016;37:576–578.
Brookes AJ, Robinson PN. Human genotype-phenotype databases: aims, challenges and opportunities. Nat Rev Genet 2015;16:702–715.
Groth KA, Gaustadnes M, Thorsen K, et al. Difficulties in diagnosing Marfan syndrome using current FBN1 databases. Genet Med 2016;18:98–102.
Collod-Béroud G, Le Bourdelles S, Ades L, et al. Update of the UMD-FBN1 mutation database and creation of an FBN1 polymorphism database. Hum Mutat 2003;22:199–208.
Landrum MJ, Lee JM, Riley GR, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 2014;42(Database issue):D980–D985.
UniProt Consortium. Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res 2014;42:D191–D198.
Lek M, Karczewski KJ, Minikel EV, et al.; Exome Aggregation Consortium. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–291.
Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 2003;31:3812–3814.
Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010;7:575–576.
Dietz HC, Pyeritz RE. Mutations in the human gene for fibrillin-1 (FBN1) in the Marfan syndrome and related disorders. Hum Mol Genet 1995;4 Spec No:1799–1809.
Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet 2013;Chapter 7:Unit7.20.
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–249.
Acknowledgements
This study was supported by the Department of Clinical Medicine, Aarhus University Hospital (http://clin.au.dk/en/) and Aarhus University (http://au.dk/en), by a PhD scholarship to K.A.G. and by the Novo Nordisk Foundation (http://www.novonordiskfonden.dk/en). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Data
(ZIP 877 kb)
Rights and permissions
About this article

Cite this article
Groth, K., Von Kodolitsch, Y., Kutsche, K. et al. Evaluating the quality of Marfan genotype–phenotype correlations in existing FBN1 databases. Genet Med 19, 772–777 (2017). https://doi.org/10.1038/gim.2016.181
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2016.181
