
Abstract
Purpose:
The Scripps Idiopathic Diseases of Man (IDIOM) study aims to discover novel gene–disease relationships and provide molecular genetic diagnosis and treatment guidance for individuals with novel diseases using genome sequencing integrated with clinical assessment and multidisciplinary case review. Here we describe the operational protocol and initial results of the IDIOM study.
Methods:
A total of 121 cases underwent first-tier review by the principal investigators to determine whether the primary inclusion criteria were satisfied, 59 (48.8%) underwent second-tier review by our clinician–scientist review panel, and 17 patients (14.0%) and their family members were enrolled.
Results:
60% of cases resulted in a plausible molecular diagnosis, and 18% of cases resulted in a confirmed molecular diagnosis. Two of three confirmed cases led to the identification of novel gene–disease relationships. In the third confirmed case a previously described but unrecognized disease was revealed. In all three confirmed cases a new clinical management strategy was initiated based on the genetic findings.
Conclusion:
Genome sequencing provides tangible clinical benefit for individuals with idiopathic genetic disease, not only in the context of molecular genetic diagnosis of known rare conditions but also in cases where prior clinical information regarding a new genetic disorder is lacking.
Genet Med 17 12, 995–1001.
Similar content being viewed by others
Introduction
The traditional approach to rare, severely disabling medical conditions frequently leaves the affected individual without a diagnosis and effective treatment. Patients with such conditions can remain ill and endure a “diagnostic odyssey” for years, which is not only difficult for such individuals and their family but can also be very cost inefficient. Many researchers have suggested the utility of genomic information for diagnosing and treating such conditions, and early evidence of the successful application of whole-exome sequencing (WES) and whole-genome sequencing (WGS) for such purposes is emerging.1,2,3,4,5,6 Of the rare likely genetic diseases that have already been described, approximately half have yet to be linked to a causal gene (referred to here as “idiopathic diseases”).7 Estimates of the total number of rare, likely genetic diseases based on the number of known disease-causing and essential genes have resulted in predictions of between 7,000 and 15,000 disorders, suggesting many rare genetic diseases have yet to be described.8 While the application of genome sequencing to the molecular genetic diagnosis of previously described rare Mendelian disorders is essentially proven, the utility of genome sequencing in novel diseases has not been systematically explored. For example, of the ~100 patients successfully diagnosed in the National Institutes of Health Undiagnosed Diseases Program (20–25% of the total enrolled), 15 cases (~3.5% of total enrolled) correspond to novel gene associations for previously described diseases, and only 2 cases (<1% of total enrolled) correspond to previously unknown diseases.9
The Scripps Idiopathic Diseases of Man (IDIOM) study was initiated in 2011. IDIOM was, in large part, modeled after the National Institutes of Health Undiagnosed Diseases Program, with a few exceptions. The primary exception is that we focus exclusively on cases that do not fit a previously described phenotype and cases in which the disorder matches a previously described phenotype and all known genetic causes of the disorder have been ruled out. In other words, only 17 of 100 cases successfully diagnosed in the Undiagnosed Diseases Program would have qualified for the IDIOM study (15 corresponding to novel gene associations for previously described diseases, 2 cases corresponding to previously unknown diseases). The application of genome sequencing to molecular genetic diagnosis in this subpopulation of individuals presents some unique challenges both in terms of the evaluation of cases and appropriateness for the IDIOM program, as well as for the ultimate return of genetic results. In this report we provide a description of IDIOM study procedures, the initial results from the first 3 years of operation, and the clinical benefit achieved by those realizing a confirmed genetic diagnosis.
Methods
Recruitment and screening
The IDIOM study (IRB-11–5723) was approved by the Scripps Institutional Review Board in 2011. Separate informed-consent forms were prepared for genome sequencing and treatment-response monitoring. Recruitment for IDIOM was done through announcements to physicians within the Scripps Health system and advocacy groups, announcements via local media,10,11 and word of mouth. Several online sources were utilized, such as ResearchMatch.org (funded by the Clinical and Translational Science Award program and hosted by Vanderbilt University), and we contacted leaders of the Undiagnosed Diseases Program, the RARE Project, and the National Organization for Rare Diseases. Scripps Health hosts a landing page for the IDIOM study that includes study criteria and coordinator contact information. The trial is also listed on ClinicalTrials.gov (http://clinicaltrials.gov/ct2/show/NCT01440218). Because initial financial support for our study was limited, we have been conservative with respect to our recruitment efforts so as not to be inundated with a large number of referrals that we would not have the resources to fund.
Inclusion criteria for the study are the following: (i) the patient has a grave or serious condition that is undiagnosed despite extensive medical and genetic evaluation—for patients with a likely clinical diagnosis, a gene panel test, at minimum, is required to rule out known genetic causes of the disorder; (ii) the patient’s condition is potentially “actionable” or amenable to treatment—this is a subjective judgment by the physician review panel that typically excludes only individuals with severe dysmorphologies; (iii) the condition seems to be genetic in origin; (iv) the patient’s anticipated life expectancy is consistent with the study timeline for sequencing; and (v) the patient has a physician champion who is willing to work with the research team and take responsibility for returning genetic results to the patient.
To be considered, a patient (or his or her referring physician) provides a short clinical summary and all available medical records. Referrals undergo an initial review by the IDIOM study coordinator. Typically, cases with complete medical records undergo another round of internal review by core study investigators, often via e-mail, and those that appear to meet inclusion criteria are forwarded for review by the IDIOM clinician–scientist review panel.
Clinician–scientist review panel
Our clinician–scientist review panel is made up of approximately 12 practicing physicians, as well as a research team consisting of bioinformatics and genetic analysis specialists, physicians who use genetics extensively in clinical practice, sequencing experts, ethicists, clinical psychologists, and research nurses. The director of the Scripps institutions review board is also a member of the panel. The clinical disciplines represented among the physician members include, but are not limited to, neurology, rheumatology, internal medicine, allergy/immunology, cardiology, medical oncology/hematology, and gastroenterology/hepatology. For a quorum we required the presence of at least five physicians and two bioinformatics or sequencing experts. Selection of cases is based on majority vote; however, in almost all instances to date, decisions to enroll a patient have been unanimous. The meetings typically last 1.5 h, and three or four cases are usually reviewed per session. We encourage and allow for, but do not require, the physician champion of patients whose cases are being reviewed to be available during the meeting (either in person or via teleconference) in order to answer questions and interface with the panel.
Consent and enrollment
Once a case has been selected by the clinician–scientist panel, our nurse study coordinator gathers consent from the patient and family members. The participants that comprise a case are usually a trio (i.e., proband, mother, and father), but occasionally other biological family members are sequenced, usually a sibling of the proband or parents.
Importantly, identification of “incidental” or “secondary” findings (i.e., genomic findings of clinical relevance that are not recognized as being associated with the presenting disease/condition) is a primary issue raised in the literature regarding patient consent for WES and WGS studies. We have deemed that best practices have yet to be determined by empirical data12 and thus return only results directly relevant to the presenting indication. If we should inadvertently discover incidental findings that could have an effect on a patient’s health, the results would be reviewed by our clinician–scientist review panel, which would adjudicate how to proceed with informing the physician champion and patient.13
For selected cases, we also ask the patient’s physician champion (usually the referring physician) to sign an agreement of participation that stipulates that he or she will commit to (i) regular interactions with the research team, (ii) acceptance of responsibility for returning genomic results to the patient, (iii) acceptance of any clinical decision making on the basis of any results provided, and (iv) completion of brief baseline and follow-up questionnaires and/or interviews pertaining to the study.
Sequence data generation, analysis, and interpretation
After cases have been selected and patients and family members have consented to participation, blood is drawn and brought to our lab at the Scripps Translational Science Institute (STSI) for sequencing of the proband and biological family members. Once parentage is confirmed, WES is performed to detect coding variants, and low-pass WGS is used to detect structural and copy-number variants. Target WES coverage is ~100× and target low-pass WGS coverage is ~5×. Other published papers have described this data-generation protocol in detail, as well as the methods we used for analysis and interpretation.14,15,16,17 If necessary, especially for insertions/deletions or variants with limited coverage in any family members, Sanger sequencing is used to confirm candidate causal variants. The theoretical target breakpoint resolution for low-pass WGS detection of copy-number variants is 200 bp given a bin size of 200–300 bp and target coverage of 4–8×.18 This resolution enables the identification of small events such as single-exon deletions. Although in practice this resolution was achieved, manual inspection of read-level WES data and low-pass WGS data was required because of false-positive variant calls. Ultimately, algorithms such as Genome STRiP, which use population-level data to account for systematic read depth biases, are necessary to improve the reliability of these results.19
Sequencing is performed in a research laboratory, and results are ultimately returned to the physician champion. Given the exploratory nature of these cases, both the return of results and the consent process include an explanation that any findings are unproven in nature and were obtained in an uncertified laboratory. Our case-selection process, with its focus on novel phenotypes and novel gene–disease relationships, eliminates from consideration individuals likely to benefit from certified laboratory tests, and we provide suggestions for the appropriate alternative commercial certified laboratories for these patients. Similarly, if a known pathogenic variant is identified, recommendations for Clinical Laboratory Improvement Amendments (CLIA) confirmation via validated tests (rather than CLIA confirmation in a certified lab without the specific validated test) are provided.
Individualized genomic report and return of results
When the results for a patient have been generated, an individualized genomic report is prepared for dissemination to our clinician–scientist review panel and the patient’s physician champion. The panel has the opportunity to raise any issues related to the case before disclosing results. In all cases, one or more consultations between members of the panel and the physician champion are arranged to allow the champion to have any questions answered and have the results verbally conveyed. In cases where a plausible diagnosis is identified, the discussion with the physician champion centers on whether the findings should change clinical management of the patient. Any new treatment offered to the patient is ultimately the treating physician’s decision. Certified genetic counselors are available if requested.
Follow-up studies
Although STSI houses the facilities required for functional studies, specialized assays are often required for appropriate functional characterization of candidate disease-causative variants. Thus, to functionally characterize findings in IDIOM, appropriate follow-up studies are tailored to the disease and gene in question, and we generally do this in collaboration with other laboratories specialized in the study of a particular gene or gene family. STSI is also uniquely facile in the use of wireless sensors. Thus, when appropriate for select patients, we design and deploy n-of-1 studies for quantitative assessment of physiologic metrics. Furthermore, we have used such approaches to objectively determine whether there has been a therapeutic response to treatment once a molecular diagnosis is made and a genomically indicated treatment initiated.
Results
Referrals and enrolled patients
In the first 3 years of the IDIOM program, 121 patient referrals were received, 59 (48.8%) of which underwent second-tier review by our clinician–scientist review panel, and 17 patients (14.0%) and their family members were enrolled. Referrals have come from more than 16 US states, and we have received several international referrals.
Demographic statistics and comparisons for referred versus reviewed versus enrolled patients are shown in Table 1 . Of the 121 patients referred to date, 31.9% were children (i.e., <18 years old), 59.4% were female, and 36.7% were referred by a physician (versus self-referred or referred by family). As shown, however, patients whose cases underwent panel review and/or were eventually enrolled were younger and more likely to be referred by a physician. Self-/family-referred subjects, if selected, were required to secure a physician champion.
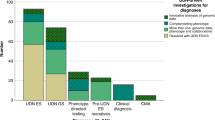
Figure 1 shows the medical specialties/phenotypes represented among the 121 patients referred to date for whom clinical information was available. By far the most common category has been neurologic disorders, encompassing 32.1% of referrals, with hematology/oncology, allergy/immunology, cardiology, and gastroenterology making up the top five broad phenotypic categories.

Phenotypic distribution of case referrals. The distribution of phenotypes referred to the Scripps Idiopathic Diseases of Man study is plotted based on the subset of 106 referrals for whom we were able to obtain complete data.
The major reasons for excluding cases have been that they meet a previously described phenotype or do not seem to be genetic. Patients whose cases meet a previously described phenotype (e.g., when the case already has a likely clinical diagnosis) are, upon exclusion, referred to an appropriate laboratory or for an appropriate test. If all known genetic causes of the previously described phenotype are ruled out, the case would be reconsidered. Exclusion of cases not likely to be genetic is usually due to one of the following situations: (i) the panel feels the condition is likely explained by an environmental insult or infectious disease, (ii) unusual circumstances surrounding the patient’s birth, for example, preterm birth or complications during birth, suggest a nongenetic developmental defect, and (iii) there is a lack of objectively documented findings either on physical examination or testing, especially subjective symptoms that cannot be connected via dysfunction of a specific biological system.
Molecular diagnosis
Detailed descriptions of each case and the findings are provided in the Supplementary Text online. Of the cases that have been processed to date, we have arrived at a plausible molecular diagnosis in approximately 60% and a confirmed molecular diagnosis in approximately 18% ( Table 2 ). A plausible molecular genetic diagnosis meets the following criteria: (i) identified variants segregate in the family and reference populations in a manner consistent with the segregation of the disease in the family and incidence of the disorder in the general population, (ii) variants influence the coding/splicing of a protein-coding gene, and (iii) the gene can be connected to the presenting phenotype through similar human disorders caused by mutation in the same gene or via genome-wide association studies, the gene can be connected to the presenting phenotype through close functional interaction with genes known to cause the presenting phenotype or a similar phenotype, or the gene can be connected to the presenting phenotype via animal studies. Of the 60% of cases meeting these criteria, we have aggressively pursued functional validation in cases where the variant is of de novo origin, further increasing confidence in the finding and maximizing the yield of downstream effort with collaborators. In two cases so far, we have completed functional and statistical confirmation of a novel gene–disease relationship via the identification of additional affected subjects with mutations in the same gene as well as functional confirmation of gene dysfunction,15,17 and in one instance a previously identified pathogenic variant was revealed. In all three cases, a new management strategy (pharmacological treatment) was initiated based on the findings, and we are currently closely monitoring these patients’ response to the therapy.
Clinical management strategy changes and their benefit
Our first case, IDIOM1, presented with a complex movement disorder, described in detail by Chen et al.; 15 it was ultimately confirmed to be a result of a gain-of-function de novo mutation in ADCY5 and potentially modified by additional mutations in DOCK3 (unconfirmed). An n-of-1-style trial to monitor nighttime abnormal movements during treatment with various compounds indicated by gain of function in ADCY5 (ropinirole, carbamazepine, tetrabenazine, and diazepam), with appropriate run-in and washout periods, was initiated. The trial was halted upon request subsequent to initiation of the first agent, diazepam, after complete resolution of nighttime myoclonic jerks. Diazepam has been shown to abrogate stress tolerance in ADCY5-null mice.20 Figure 2 shows a dramatic and sustained resolution of abnormal movements after initiation of diazepam treatment, as captured by a movement-tracking device.

Tracking of therapy response in IDIOM1. Actigraphy-based motion tracking demonstrates a dramatic and sustained decrease in nighttime myoclonic jerks caused by a gain-of-function mutation in ADCY5. Day 0–6 represents the last week of a 3-week run-in period to wash out previous therapies. Diazepam was initiated on day 6 (arrow), with a dramatic reduction in tremors sustained for over 2 months. Tremors are defined as movement magnitude >0 sustained for longer than 60 s.
Patient IDIOM9 presented with a complex seizure disorder with drop attacks, described in detail by Torkamani et al.17 and ultimately confirmed to be due to a de novo mutation in potassium channel KCNB1 that changed ion selectivity. As a result, in order to carefully control potassium concentrations, the subject was placed on a specialized diet and kept hydrated. Thus, a change in the clinical management strategy was initiated, with unconfirmed clinical benefit. Unfortunately, no particular antiepileptic drug is indicated from the genetic results, and comparison with patients with a similar underlying genetic cause of epilepsy did not reveal a specific and efficacious therapeutic strategy (ref. 21 and personal communication S. Berkovic, MD). An anecdotal reduction in drop attacks has been noted by the treating physician, although longer follow-up is required to confirm a sustained benefit.
Patient IDIOM15 presented with hypertrichotic osteochondrodysplasia (excess hair growth on the scalp, forehead, and face) due to a gene defect that was ultimately attributed to a known pathogenic de novo ABCC9 mutation. Dominant missense mutations in ABCC9 cause gain-of-function channel opening and implicate several channel blockers as potential therapeutic avenues.22 Treatment modifications have been initiated, and long-term follow-up is required to confirm a sustained benefit.
Discussion
The IDIOM study aims to discover novel gene–disease relationships and provide molecular genetic diagnosis and treatment options for individuals with idiopathic diseases using genome sequencing integrated with clinical assessment and multidisciplinary case review. The primary difference from similar programs is our exclusive focus on novel gene–disease relationships. In fact, candidates with phenotypes similar to ones previously described must have known genes potentially mediating their phenotype ruled out before being considered as an IDIOM subject. Given our protocol’s exclusive focus on novel diseases and novel gene–disease relationships, it is remarkable that we demonstrate an initial rate of novel and confirmed genetic discoveries similar to that reported by programs focused on standard molecular genetic diagnosis via genome sequencing (20–25%).3,4,9,23 Although the volume, rate, and efficiency at which novel gene–disease relationships can be confirmed are lower than those achievable by standard molecular genetic diagnosis via genome sequencing, our results suggest that genome sequencing has the potential to be at least as efficacious in providing genetic diagnoses for individuals with previously undescribed disease as it is for individuals with known Mendelian disorders, with an upper limit to the diagnostic yield of ~60% (±23–95% confidence interval), as informed by our plausible findings. However, we must acknowledge that a direct comparison between the diagnostic rate of these programs cannot be made because the rates depend heavily on ascertainment of the cohort in question.
The conversion of novel, plausible gene–disease relationships to confirmed and validated genetic diagnoses should increase the yield of clinical genome sequencing programs above the current 20–25% diagnostic rate. In fact, individuals sequenced at clinical genome sequencing centers have initiated contact with the IDIOM investigators subsequent to publication of our confirmed novel gene–disease relationships, bringing to light the fact that some individuals do not achieve a genetic diagnosis because of a lack of recognition of the causal variant in genome sequence data by those reporting the results rather than any deficit in the technical identification of the causal variant. For example, we have received inquiries from two individuals with negative clinical exome results who had the same de novo ADCY5 mutation as that described in patient IDIOM1; this was reported—correctly, at the time of the test—as a variant of unknown significance in the clinical exome report. The relative contribution of deficits in technical variant identification versus deficits in knowledge of gene–disease relationships to the lack of molecular genetic diagnosis in those ~75% of individuals not receiving a genetic diagnosis after clinical genome sequencing remains to be determined. Ultimately, the contribution of novel gene–disease relationships to increasing the yield of clinical genome sequencing requires conversion of plausible findings to confirmed genetic diagnoses through the identification of additional cases. This has been difficult to achieve but may be facilitated through services such as Matchmaker Exchange (http://www.matchmakerexchange.org). Comprehensive and accessible collection of phenotypic characteristics is key to achieving this goal.
In our study, all confirmed molecular genetic diagnoses have led to a tangible clinical benefit above and beyond the utility achieved simply by ending a diagnostic odyssey. However, there have been notable challenges in the context of this project. For example, we often have families request other information, frequently referred to as secondary or “incidental” findings, from their genomic analysis, and the research team has engaged in substantial discussion about whether findings that do not pertain to a patient’s presenting condition should be assessed and reported back to the physician and/or patient. At this time, given that sequencing is performed in an uncertified laboratory for a prespecified purpose, we do not return information that is not directly applicable to the presenting condition. We have also observed wide variability in our physician champions’ knowledge of and comfort with genomic information, which has a major influence on the amount of information these clinicians share with patients and the ultimate clinical utility of any findings. A multidisciplinary clinician–scientist review panel has been essential to supporting physician champions as well as selecting cases most likely to benefit from genome sequencing. Finally, the transmission of information back to physician champions, clearly indicating the suggestive nature of the majority of our findings, has been essential to managing expectations of the physician champions and ultimately the enrolled subjects.
An important area of improvement, especially for this program with no specific disease focus, is to objectively link the symptoms presented by study subjects to previously described conditions. Any physician–scientist review panel is unlikely to represent specialties covering the wide variety of conditions referred to the program; thus, similarities to previously described conditions (as exemplified by our hypertrichotic osteochondrodysplasia case) may be missed. In a more comprehensive program, spanning molecular genetic diagnosis of undiagnosed disease and discovery of novel gene–disease relationships in idiopathic disease, automated systems for phenotype matching can identify both known genetic conditions and gene–disease relationships that should be considered for confirmatory molecular diagnosis, as well as grounding for exploration of novel gene–disease relationships through implicated biological processes and genetic networks mediating those processes. We believe this could provide a model for the unbiased application of genome sequencing across all rare genetic disorders, known and unknown.
Disclosure
A.A.S.-V.Z., E.J.T., A.T., and N.J.S. declare they are cofounders and equity holders of Cypher Genomics, Inc. The other authors declare no conflict of interest.
References
Jacob HJ, Abrams K, Bick DP, et al. Genomics in clinical practice: lessons from the front lines. Sci Transl Med 2013;5:194cm5.
Worthey EA, Mayer AN, Syverson GD, et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med 2011;13:255–262.
Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 2014;312:1870–1879.
Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 2013;369:1502–1511.
de Ligt J, Willemsen MH, van Bon BW, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 2012;367:1921–1929.
Srivastava S, Cohen JS, Vernon H, et al. Clinical whole exome sequencing in child neurology practice. Ann Neurol 2014;76:473–483.
Boycott KM, Vanstone MR, Bulman DE, MacKenzie AE. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat Rev Genet 2013;14:681–691.
Cooper DN, Chen JM, Ball EV, et al. Genes, mutations, and human inherited disease at the dawn of the age of personalized genomics. Hum Mutat 2010;31:631–655.
Gahl WA, Markello TC, Toro C, et al. The National Institutes of Health Undiagnosed Diseases Program: insights into rare diseases. Genet Med 2012;14:51–59.
Robbins G. The search for what’s wrong with Lilly. San Diego Union-Tribune 2013.
Yong E. “We gained hope.” The story of Lilly Grossman’s genome. National Geographic 2013.
Tabor HK, Berkman BE, Hull SC, Bamshad MJ. Genomics really gets personal: how exome and whole genome sequencing challenge the ethical framework of human genetics research. Am J Med Genet A 2011;155A:2916–2924.
Mayer AN, Dimmock DP, Arca MJ, et al. A timely arrival for genomic medicine. Genet Med 2011;13:195–196.
Belani R, Oliveira G, Erikson GA, et al. ASXL1 and DNMT3A mutation in a cytogenetically normal B3 thymoma. Oncogenesis 2014;3:e111.
Chen YZ, Friedman JR, Chen DH, et al. Gain-of-function ADCY5 mutations in familial dyskinesia with facial myokymia. Ann Neurol 2014;75:542–549.
Brownstein CA, Beggs AH, Homer N, et al. An international effort towards developing standards for best practices in analysis, interpretation and reporting of clinical genome sequencing results in the CLARITY Challenge. Genome Biol 2014;15:R53.
Torkamani A, Bersell K, Jorge BS, et al. De novo KCNB1 mutations in epileptic encephalopathy. Ann Neurol 2014;76:529–540.
Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res 2011;21:974–984.
Handsaker RE, Korn JM, Nemesh J, McCarroll SA. Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nat Genet 2011;43:269–276.
Kim KS, Han PL. Mice lacking adenylyl cyclase-5 cope badly with repeated restraint stress. J Neurosci Res 2009;87:2983–2993.
Muona M, Berkovic SF, Dibbens LM, et al. A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy. Nat Genet 2015;47:39–46.
Harakalova M, van Harssel JJ, Terhal PA, et al. Dominant missense mutations in ABCC9 cause Cantú syndrome. Nat Genet 2012;44:793–796.
LaDuca H, Stuenkel AJ, Dolinsky JS, et al. Utilization of multigene panels in hereditary cancer predisposition testing: analysis of more than 2,000 patients. Genet Med 2014;16:830–837.
Acknowledgements
The authors thank the patients and their families for their cooperation. This work is supported by Scripps Genomic Medicine, a National Institutes of Health National Center for Advancing Translational Studies Clinical and Translational Science Award (5 UL1 RR025774) to STSI, as well as funding from the Shaffer Family Foundation and the Anne and Henry Zarrow Foundation. Further support is from National Institutes of Health grant U01 HG006476 (to A.T.).
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Text
(DOC 118 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Bloss, C., Zeeland, AV., Topol, S. et al. A genome sequencing program for novel undiagnosed diseases. Genet Med 17, 995–1001 (2015). https://doi.org/10.1038/gim.2015.21
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2015.21
Keywords
This article is cited by
-
Real-world economic evaluation of prospective rapid whole-genome sequencing compared to a matched retrospective cohort of critically ill pediatric patients in the United States
The Pharmacogenomics Journal (2022)
-
Comparative analysis of whole-genome sequencing pipelines to minimize false negative findings
Scientific Reports (2019)
-
SG-ADVISER mtDNA: a web server for mitochondrial DNA annotation with data from 200 samples of a healthy aging cohort
BMC Bioinformatics (2017)
