
Abstract
Purpose:
Four mitochondrial metabolic liver enzymes require bicarbonate, which is provided by the carbonic anhydrase isoforms VA (CAVA) and VB (CAVB). Defective hepatic bicarbonate production leads to a unique combination of biochemical findings: hyperammonemia, elevated lactate and ketone bodies, metabolic acidosis, hypoglycemia, and excretion of carboxylase substrates. This study aimed to test for CAVA or CAVB deficiencies in a group of 96 patients with early-onset hyperammonemia and to prove the disease-causing role of the CAVA variants found.
Methods:
We performed CA5A and CA5B sequencing in the described cohort and developed an expression system using insect cells, which enabled the characterization of wild-type CAVA, clinical mutations, and three variants that affect functional residues.
Results:
In 10 of 96 patients, mutations in CA5A were identified on both alleles but none in CA5B. Exhibiting decreased enzyme activity or thermal stability, all CAVA mutations were proven to cause disease, whereas the three variants showed no relevant effect.
Conclusion:
CAVA deficiency is a differential diagnosis of early-onset and life-threatening metabolic crisis, with hyperammonemia, hyperlactatemia, and ketonuria as apparently obligate signs. It seems to be more common than other rare metabolic diseases, and early identification may allow specific treatment of hyperammonemia and ultimately prevent neurologic sequelae.
Genet Med 18 10, 991–1000.
Similar content being viewed by others


Introduction
Bicarbonate (HCO3−) is the predominant extracellular buffer and is essential for maintaining pH homeostasis throughout the human body. Carbonic anhydrases (CAs), a family of zinc metalloenzymes, catalyze the reversible hydration of CO2 ( ).1 These enzymes are involved in processes such as respiration, renal acidification, bone resorption, formation of aqueous humor and cerebral fluid, gastric secretion, gluconeogenesis, lipogenesis, and ureagenesis in humans.2 The 14 isoenzymes known in humans differ in tissue-specific expression, subcellular localization, kinetic parameters, and susceptibility to various inhibitors; for example, CAs I, II, III, VII, and XIII are located in the cytoplasm; CAs IV, IX, XII, and XIV are membrane bound; CAs VI, X, and XI are secreted; and CAVA and VB are located in the mitochondria (http://www.proteinatlas.org).
).1 These enzymes are involved in processes such as respiration, renal acidification, bone resorption, formation of aqueous humor and cerebral fluid, gastric secretion, gluconeogenesis, lipogenesis, and ureagenesis in humans.2 The 14 isoenzymes known in humans differ in tissue-specific expression, subcellular localization, kinetic parameters, and susceptibility to various inhibitors; for example, CAs I, II, III, VII, and XIII are located in the cytoplasm; CAs IV, IX, XII, and XIV are membrane bound; CAs VI, X, and XI are secreted; and CAVA and VB are located in the mitochondria (http://www.proteinatlas.org).
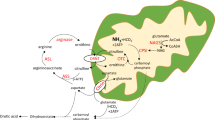
Human CAVA is a 267–amino acid mitochondrial enzyme expressed exclusively in the liver, kidneys, and skeletal muscle.3 It provides bicarbonate to four hepatic mitochondrial enzymes involved in essential metabolic pathways: carbamoyl phosphate synthetase 1 (CPS1; the first step of the urea cycle), pyruvate carboxylase (gluconeogenesis), propionyl-CoA carboxylase, and 3-methylcrotonyl-CoA carboxylase (branched-chain amino acid catabolism) ( Figure 1a ). The encoding gene CA5A (OMIM 114761) spans ~50 kb, contains 7 exons and 6 introns, and maps to 16q24.2 (refs. 4 and 5). Of all human CAs, CAVB is the only other isoform expressed in mitochondria, but it has a wider tissue distribution, including the liver.3 CA5B (OMIM 300230) spans ~50 kb as well, but it maps to Xp22.2 (ref. 6).

Biochemical pathways using bicarbonate produced by carbonic anhydrase VA (CAVA) and mapping of CAVA clinical mutations. (a) Dotted lines link CAVA-produced bicarbonate (orange) with the bicarbonate-dependent carboxylases (in italics and blue). Enzymes: CPS1, carbamoyl phosphate synthetase 1; PC, pyruvate carboxylase; PCC, propionyl CoA carboxylase; 3MCC, 3-methylcrotonyl CoA carboxylase. Metabolites: AS, argininosuccinate; 3MCCoA, 3-methylcrotonyl CoA; 3MGCoA, 3-methylglutaconyl CoA; AcCoA, acetyl CoA; αKG, α-ketoglutarate; OAA, oxaloacetate; SUC, succinyl CoA; Glu, glutamate; Gln, glutamine; Sac, saccharopine; Lys, lysine; ASP, aspartate; PEP, phosphoenolpyruvate; PYR, pyruvate; LAC, lactate. (b) Human wild-type CA5A gene representation with predicted mitochondrial targeting sequence (MTS) and predicted functional residues for zinc binding (blue), active site (green), and substrate binding (cyan). Clinical mutations are shown in pink. p.Ser233Pro, p.Lys185Lys, leading to the deletion of exon 4 and the deletion of exon 6, were published in ref. 8. (c) Mouse CAVA structure (Protein Data Bank code 1DMX) in the cartoon representation is gray. Functional residues are in the stick representation (C atoms are colored as in (b), and O atoms are red). Residues harboring the missense mutations studied here are shown as pink spheres, and the regions corresponding to exons 4 (residues 154–186) and 6 (residues 207–258) are colored in dark and light pink, respectively. Polymorphism p.Pro273Leu is represented as a yellow sphere and labeled. Polymorphisms p.Asn45Lys and p.Asn46Lys are not represented since the most N-terminal part of the protein is not included in the crystal structure. Zinc is represented as a small, dark gray sphere and labeled.
The liver disease CAVA (CAVA) deficiency (OMIM 615751) is a rare autosomal-recessive inborn error of metabolism (IEM)7 that results in defective hepatic bicarbonate production. This novel disorder was recently described in four patients8 exhibiting lethargy caused by neonatal or infantile hyperammonemia, combined with elevated blood lactate and urine ketone bodies, metabolic acidosis, hypoglycemia, and urinary excretion of carboxylase substrates and related metabolites.8 On the other hand, no human disease has been attributed to a defect in CAVB.
Until 2014, the only known human disease attributable to the deficiency of a CA was CAII deficiency. CAII is found in the cytosol of essentially every tissue or organ,2 and its deficiency is associated with osteopetrosis, renal tubular acidosis, and cerebral calcifications.9,10 Bone marrow transplantation has been used to attenuate osteopetrosis and cerebral calcification,11 and sodium bicarbonate has been used to treat renal tubular acidosis12 in these patients. By contrast, deficiency of CAI, a cytosolic isoform highly expressed in erythrocytes, does not produce a clinical phenotype13 in either humans or in pigtail macaques.14,15
There exist few animal models with defective CAs. One is the CAII-deficient mouse, which only partly recapitulates the human disease: it exhibits only renal tubular acidosis and cerebral calcifications, not the prominent osteopetrosis found in humans with this disease.16 Promising results have been obtained using gene therapy in CAII-deficient mice, which was able to restore the ability to acidify urine after oral administration of ammonium chloride.17 The mouse model lacking Car5A (murine homologue to human CAVA), like humans, exhibited life-threatening hyperammonemia, whereas the Car5B (murine homologue to human CAVB) knockout mice had no clinical phenotype.18 Interestingly, double-knockout (Car5A−/−, Car5B−/−) mice developed even more severe hyperammonemia than the Car5A knockout mice, suggesting that even though both Car5A and Car5B seem to contribute to ureagenesis in mice, Car5A has a predominant role.18
Given the amenability to treatment and therefore the prevention of irreversible brain damage, our group was motivated to investigate further the effect of impaired hepatic bicarbonate production, at both the clinical and biochemical levels, as a cause of hyperammonemic encephalopathy. Our aims were, first, to test for CAVA deficiency in patients with “isolated” neonatal-onset hyperammonemia, in whom the biochemical profile thus was not indicative of any other urea cycle disorder (UCD) or organic aciduria. To do so, we sequenced CA5A in a group of 96 patients who were initially considered to suffer from a proximal UCD but in whom no mutations in NAGS and CPS1 were found. In addition, to rule out a CAVB defect, we sequenced CA5B. Third, to prove the disease-causing role of all detected CA5A mutations, we developed an expression system based on insect cells, already proven valid by other groups to express other CA isoforms19,20 and by our group to study another liver mitochondrial enzyme, CPS1.21,22,23 We expressed the recombinant wild-type CAVA, all published and newly identified mutations, and three non-disease-associated variants, and we tested their enzyme activity and thermal stability. Finally, we described the clinical and biochemical features of the patients with CAVA deficiency.
Materials and Methods
Patients
This study was carried out in a group of 96 patients (48 male, 46 female, 2 with gender unknown to us) referred to us between 2008 and 2014 from 50 metabolic centers worldwide. We received 313 samples from index patients for NAGS and/or CPS1 mutation analysis; of these, 15 and 83 were positive for N-acetyl-glutamate synthase (NAGS) and CPS1 deficiencies, respectively. Of the negative samples, we include here only those negative for NAGS and CPS1 genes. NAGS testing was done by single-exon amplification and DNA sequencing.24 CPS1 testing was done by RNA sequencing, in most cases using stimulated lymphocytes25 and in a few patients using cultured fibroblasts.26 Samples of this group were received from many European countries and from Australia, Brazil, Israel, Kuwait, Saudi Arabia, and Tunisia. The majority of patients (n = 60) were referred from large centers that sent samples from several patients (between 2 and 9 samples), whereas 36 patients were single referrals. We thus relied on clinical and biochemical information from many different places.
All patients studied were initially suspected to suffer from a proximal UCD since they presented with symptomatic hyperammonemia in the absence of increased urinary orotic acid. Most of the patients suffered an initial hyperammonemic crisis during the first days of age, but few patients presented at a later age during infancy or childhood ( Tables 1 and 2 ).
Molecular genetic analysis
In the entire group, standard sequence analysis of coding exons, including flanking intronic regions of CA5A and CA5B, was performed. If not provided, genomic DNA was extracted from peripheral blood leukocytes or stored skin fibroblasts. Genomic DNA was amplified by polymerase chain reaction, followed by agarose gel electrophoresis and direct sequencing according to standard protocols using the BigDye Terminator Cycle Sequencing kit version 1.1 and an ABI 3130 genetic analyzer (Applied Biosystems by Life Technologies Europe BV, Zug, Switzerland). Because of the highly homologous pseudogene sequences, the primers used were designed to be unique at their 3′ end (Supplementary Table S1 online). Raw sequencing data were analyzed using the SeqPatient module of SEQUENCE PILOT software (JSI Medical Systems, Ettenheim, Germany) using the following reference sequences: CA5A Ensembl ENSG00000174990 and ENST00000309893; CA5B Ensembl ENSG00000169239 and ENST00000318636. Single-nucleotide polymorphism variants were excluded by blasting the sequencing data against the Ensemble platform. Nomenclature of mutations follows the recommendations of the Human Genome Variation Society (http://www.hgvs.org/mutnomen) and was checked using the online software Mutalyzer (https://mutalyzer.nl/name-checker). Mutation analysis was performed after written informed consent of the legal guardian was obtained. The study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki. To confirm segregation of the mutations, parental samples were analyzed whenever available.
Amino acid conservation (Supplementary Table S2 online) was determined by ClustalW sequence alignment of CAVA from 66 species or the other 13 human CAs. The frequency of the mutations was analyzed using the Exome Aggregation Consortium browser (http://exac.broadinstitute.org/about) (Supplementary Table S2 online). The disease-causing probability of all variants was tested by the prediction servers PolyPhen-2 (ref. 27) and MutPred.28
Enzyme activity assay
CA activity assay was performed using a version adapted from the changing pH indicator method developed by Maren,29 as described in the “Enzymatic Assay of Carbonic Anhydrase for Wilbur-Anderson Units” document from Sigma (http://www.sigmaaldrich.com/technical-documents/protocols/biology/enzymatic-assay-of-carbonic-anhydrase.html). It was based on the monitoring of pH variation resulting from the catalyzed conversion of CO2 to bicarbonate. The assay was performed at 0–3 °C in a 24-well plate, which contained a pH-meter electrode and a magnet (for mixing), and which was placed in an ice-water bath on top of a magnetic agitator. Ice-cold CO2-saturated water (1.6 ml) was added to 2 ml of 20 mmol/l Tris-SO4 buffer (pH 8.3). The CO2-saturated solution was prepared by adding dry ice chips into ice-cold water, keeping it bubbling for 15 min before starting the experiment. To ensure that the solution remained saturated with CO2 throughout the whole experiment, dry ice chips were added regularly to maintain the bubbling. Immediately after, 15–75 µg of wild-type or mutant CA were added to the buffer–substrate solution, and a stopwatch was started simultaneously. For the blanks, an equivalent volume of buffer was added to the test tube. The time required for the solution to change from pH 8.3 to 6.3 was recorded using pH Meter 744 (Metrohm, Herisau, Switzerland). Enzyme activity was calculated in Wilbur-Anderson units according to the formula (T0 − T)/T, where T0 (blank) and T (catalyzed reaction by CA) are the time (in seconds) required for the pH to change from 8.3 to 6.3.
Results
Patients with CAVA and mutations
Among the group of 96 patients, we identified CA5A mutations in 10 patients ( Table 1 ). These patients presented with a unique combination of biochemical findings, including hyperammonemia (in all patients; range: 238–1,150 μmol/l), elevated lactate (in all patients; range: 4.8–15 mmol/l), and elevated ketone bodies in urine (in all patients) ( Table 2 ). Glutamine and glucose concentrations varied among patients: between normal limits to 2,606 μmol/l, and between normo- and hypoglycemia (1.9 mmol/l), respectively ( Table 2 ). Metabolic acidosis and urinary excretion of carboxylase substrates and related metabolites were also observed in variable ranges ( Table 2 ). For a full picture of the currently known patients with CAVA deficiency, we also include the previously published8 patients (numbers 3, 5, 6, and 9 in Tables 1 and 2 ). Seven of the 10 newly described patients and 2 already described in ref. 8 presented with only one initial hyperammonemic crisis, after which they remained stable, even after tapering of treatment ( Table 1 ). However, three newly described patients suffered a second crisis, which was milder than their first one ( Tables 1 and 2 ); the two siblings already described in ref. 8 presented with multiple crises during illness, albeit milder the second and third times. Some patients were treated with N-carbamyl-l-glutamate (NCG) during their initial crisis, either alone or in combination with other measures, and seemed to respond positively ( Table 1 ), with rapid reduction of ammonia.
Ninety percent of the newly described patients came from the Indian subcontinent (India, Pakistan, or Bangladesh), and one patient was Caucasian (Turkish descent). In terms of variants identified, the nonsense mutation p.W41* ( Figure 1b ), identified in a Turkish patient, is not expected to lead to nonsense-mediated decay of mRNA because it does not fulfill respective criteria,30 but it will likely lead to a substantially shortened and thus inactive protein. The novel change p.Glu241Lys ( Figure 1b , c ), found in two independent patients originating in Bangladesh and Pakistan, respectively, was predicted by in silico analysis to cause disease (Supplementary Table S2 online). The splice-site deletion of exon 3, found in one Indian patient, probably results in the skipping of exon 3, leading to a frame shift p.(Gly114Alafs*53) ( Figure 1b ). The remaining six patients, all from the Indian subcontinent ( Table 1 ), carry an in-frame deletion of either exon 4 (patient 4) or 6 (patients 10–14) ( Figure 1b , c ); thus, because of the large protein region deleted and because of the removal of catalytic residues ( Figure 1b , c ), these mutations are expected to cause enzyme inactivation, if not lead to protein misfolding and degradation. On the other hand, prediction servers made predictions of benignity for the two most frequent polymorphisms (p.Asn45Lys, rs77325391, and p.Asn46Lys, rs74041853), included here as negative controls (Supplementary Table S2 online). By contrast, p.Pro237Leu, the third non-disease-associated variant included in this study ( Figure 1b , c ), was predicted to be probably damaging (Supplementary Table S2 online).
Expression, isolation, and stability of recombinant wild-type CAVA, its clinical mutants, and three known polymorphisms
To characterize all the published and newly identified CA5A variants, we introduced CA5A constructs carrying wild-type or mutant (primers are provided in Supplementary Table S3 online) complementary DNA into Sf9 insect cells to express CAVA proteins using the baculovirus/insect cell expression system (Supplementary Materials and Methods online). Human CAVA is produced in the liver as a precursor, preceded by an N-terminal 38–amino acid mitochondrial signaling peptide, which is cleaved upon entry into the mitochondria. Thus, for expression of recombinant CAVA, we used the complementary DNA of mature human CAVA (the functional form in vivo) without the N-terminal 38 codons, which were replaced by a 28-codon N-terminal His6 tag to facilitate purification. Since the presence of this tag drastically lowered enzyme activity (data not shown), it was removed using tobacco etch virus protease treatment ( Figure 2a ) after protein purification. The three non-disease-associated variants (p.Asn45Lys, p.Asn46Lys, and p.Pro237Leu) studied as controls and the two missense clinical mutations (p.Ser233Pro and p.Glu241Lys) did not hamper CAVA expression, solubility, or enzyme purification ( Figure 2a ). However, the mutations carrying a deletion of exon 4 or a deletion of exon 6 exhibited lower enzyme yields ( Figure 2a ) apparently as the result of poor polypeptide production and gross misfolding; only a small fraction of the expressed protein could be cleaved with the tobacco etch virus.

Production, thermal stability, and activity of wild-type (WT), polymorphisms, and clinical mutations of CAVA. WT+ and WT− indicate the presence and absence, respectively, of the N-terminal His6 tag in the WT enzyme. In all polymorphisms and mutant forms, the N-terminal His6 tag has been removed by tobacco etch virus (TEV) treatment. Polymorphisms are labeled in gray and underlined. (a) Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) (15% polyacrylamide, Coomassie blue staining) of purified human recombinant CAVA, either WT or carrying the indicated mutations. St, protein markers (PageRuler Plus prestained protein; Thermo Scientific), with masses indicated in kilodaltons. The black arrowhead indicates the position of the tag-free CAVA band of the WT form, polymorphisms, and missense mutations. The pink arrowhead indicates the position of the tag-free CAVA band of the enzyme forms carrying deletions of exon 4 or 6, respectively. (b and c) Thermostability of CAVA forms. (b) The change in Tm values (ΔTm, compared with WT−) for each CAVA mutant. Error bars depict SEM from at least six measurements. *Statistically different (P < 0.05, Mann–Whitney test). (c) Differential scanning fluorimetry melting curves of WT CAVA and the three most unstable clinical mutations. (d) Enzyme activity per milligram of pure CAVA, expressed relative to the corresponding activity of the pure WT enzyme (438.4 U/mg). Error bars depict the SEM from at least six measurements. *Statistically different (P < 0.05, Mann–Whitney test).
Mutations harboring deletions of exons 4 or 6 render CAVA inactive
In addition to the decreased enzyme production observed in the two enzyme forms carrying the deletions of exons 4 or 6, these mutants exhibited decreased thermal stability at ~3 C and ~4 °C, respectively ( Figure 2b , c and Table 3 ), relative to wild-type CAVA. In both cases the decreased stability might be explained by the elimination of a large fraction of the enzyme (32 residues in the case of the mutant with the deletion of exon 4, and 52 residues in the case of the mutant with the deletion of exon 6, which correspond to ~12 and ~19% of the enzymes, respectively) ( Figure 1c ). Moreover, these two mutations render CAVA inactive ( Figure 2d and Table 3 ), confirming their disease-causing nature. Exon 4 contains the zinc binding site and the active site of the enzyme, whereas the substrate binding sites are located in exon 6 ( Figure 1b , c ). Therefore these mutations should inactivate CAVA by eliminating these key residues for the enzyme’s activity.
Clinical missense mutations p.Ser233Pro and p.Glu241Lys decrease enzyme activity and thermal stability
Two clinical missense mutations, p.Ser233Pro and p.Glu241Lys, decrease enzyme-specific activity by ~50 and ~75% ( Figure 2d and Table 3 ), respectively; this is probably explained by the fact that these two mutations affect residues mapping close to the substrate binding site ( Figure 1b , c ). In addition to the decreased specific activity ( Figure 2d and Table 3 ), mutant p.Ser233Pro lowered the Tm(melting temperature) by ~6 °C ( Figure 2b , c and Table 3 ), leading to the expectation of an important reduction in enzyme level in the tissue. These results corroborate those published in ref. 8, where mutant p.Ser233Pro was proven to be unstable. Similarly, the newly-described mutation p.Glu241Lys, apart from decreasing enzyme activity by ~75% ( Figure 2d and Table 3 ), also decreased the Tm by ~2 °C ( Figure 2b and Table 3 ). The decreased thermal stabilities may result in shorter half-lives of these two mutant forms in liver mitochondria; this fits the clinical course with metabolic decompensations triggered by febrile episodes, which is common to other IEMs as well (e.g., medium-chain acyl-CoA dehydrogenase deficiency31). Thus the disease-causing nature of these missense mutations would be explained by the combination of decreased residual enzyme activity as well as decreased thermal stability, in keeping with the clinical course of recurrent fever-induced decompensations during early childhood.
p.Asn45Lys, p.Asn46Lys, and p.Pro237Leu are variants without detectable functional consequences
The two most frequent polymorphisms (p.Asn45Lys and p.Asn46Lys), located in the N-terminal region of the enzyme, showed no substantial effect on CAVA activity ( Figure 2d and Table 3 ) or thermal stability ( Figure 2b and Table 3 ). This lack of effects can be rationalized by the fact that they map to residues located in the very N-terminal region of the enzyme, far away from the active site, and that they affect residues that vary among other CAs (Supplementary Table S2 online). The other non-disease-associated variant p.Pro237Leu decreased CAVA-specific activity by ~30% ( Figure 2d and Table 3 ), probably because it is involved in substrate binding ( Figure 1c ). However, this slight reduction in specific activity is expected to have no functional consequences in vivo.
Discussion
The liver disease CAVA deficiency was described in 2014 as a human IEM7 in four patients.8 In this work we describe 10 more patients with CAVA deficiency who were identified among a group of 96 patients with unexplained hyperammonemia, suggesting that this disease may be more common than the rare forms of UCDs such a NAGS deficiency (estimated incidence: <1:2,000,000).32
Detailed review of the phenotypes of the 10 patients identified here supports previous reports of CAVA deficiency; this IEM presents with unique biochemical findings during the neonatal period and infancy, including lethargy, poor feeding, and vomiting caused by hyperammonemia, combined with elevated blood lactate and urinary ketone bodies, metabolic acidosis, hypoglycemia, and excretion of carboxylase substrates and related metabolites. Not all of these biochemical findings were present in all patients, but hyperammonemia, hyperlactatemia, and ketonuria seem to be obligatory ( Table 2 ). These biochemical observations are consistent with the impairment of all four enzymes that use as a substrate the bicarbonate produced by CAVA in the liver mitochondria: CPS1, which converts waste nitrogen in the form of ammonia, bicarbonate, and adenosine triphosphate into carbamoyl phosphate in the first step of the urea cycle; pyruvate carboxylase, which links glycolysis with the Krebs cycle, by converting pyruvate to oxaloacetate, an essential substrate for the Krebs cycle; and propionyl-CoA carboxylase and 3-methylcrotonyl-CoA carboxylase, which are involved in branched-chain amino acid catabolism ( Figure 1a ). In particular, the degree of hyperammonemia in CAVA-deficient patients during metabolic crisis varied between 238 and 1,150 μmol/l, similar to other UCDs, and these patients accumulated ketone bodies and organic acids such as 3-OH propionic acid and exhibited variable levels of metabolic acidosis characteristic of secondary propionyl-CoA carboxylase and 3-methylcrotonyl-CoA carboxylase deficiencies.
Indicating a possible but yet unproven common ancestry, 43% of the patients carried the homozygous CA5A exon 6 deletion. Indeed, all these patients come from the Indian subcontinent (either India or Pakistan), and in some cases, consanguinity is known ( Table 1 ). In contrast to this hypothesis, patient 11 ( Table 1 ), in spite of coming from India, had a Hindu background rather than an Islamic one; thus the exon 6 deletion probably originated genuinely in separate communities. Likewise, patient 14 ( Table 1 ), despite being Pakistani, was born to nonconsanguineous parents. The issue of consanguinity also has to be considered as a contributing factor for the neurological symptoms in patient 13, which may be unrelated to CAVA deficiency.
NCG, although not approved as a treatment for CAVA deficiency, seemed to be beneficial for these patients ( Table 1 ). We postulate the following explanation for its therapeutic mechanism: since propionyl-CoA carboxylase cannot perform its reaction properly because of the absence of HCO3−, propionyl-CoA, a potent inhibitor of NAGS,33 accumulates. The decreased production of NAG, an essential activator of CPS1, in addition to the low concentration of its substrate bicarbonate, hampers CPS1 activity and thus ammonia detoxification by the urea cycle. The resulting secondary hyperammonemia exhibited by these patients is amenable to NCG via an increase in CPS1 activation. It must be acknowledged, however, that pyruvate carboxylase deficiency also contributes to hyperammonemia, albeit to a lesser extent given the absence of an increase in α-ketoglutarate and the relatively mild increase of Krebs cycle intermediates; thus this life-threatening symptom may strongly yet incompletely respond to NCG. In conclusion, CAVA deficiency should be considered as a treatable IEM given its amenability to NCG, in addition to extracorporeal detoxification in the case of severe hyperammonemia, as well as standard treatment of the acute metabolic crisis, which includes increased administration of calories and fluids and decreased protein intake and intravenous lipids.
A remarkable observation was that ~65% of the patients with CAVA described presented only one initial crisis, after which they remained stable, even after tapering treatment. In the remaining ~35%, the subsequent crises were triggered by catabolism and febrile episodes, and all these crises were less severe than the initial ones. A suggested explanation for this phenomenon is that the CAVB isoform, the only other CA expressed in liver mitochondria, undergoes a maturation effect,27 by which the expression of functional CAVB develops during early infancy. In patients with defective CAVA, CAVB would not be sufficiently present or would be immature at the moment of birth, explaining the initial crisis, but would become functional over time, stabilizing the CAVA-deficient patient and attenuating further metabolic crisis.8
The expression system presented here provides evident correlations between the clinical mutations and the phenotypes observed in the patients, enabling the understanding of the degree of severity of different mutations. Indeed, the lack of functional effects of the three non-disease-associated variants studied here, and the severity of the effects (decreased enzyme activity and/or thermal stability) of the four clinical mutations analyzed, indicate that the experimental studies with recombinant CAVA mutants expressed “in vitro” validate disease-causing mutations.
In summary, our study further confirms CAVA deficiency as a novel differential diagnosis of neonatal and early-childhood hyperammonemia, and it seems more common than other rare IEMs such as NAGS deficiency. It is even possible that patients who were labeled with the exclusion diagnosis of transient hyperammonemia of the newborn could have been affected by CAVA deficiency. This novel hepatic disease exhibits a unique biochemical fingerprint including hyperammonemia, elevated blood lactate and urinary ketone bodies, metabolic acidosis, hypoglycemia, and excretion of carboxylase substrates and related metabolites. The metabolic crisis can be very severe at presentation and should be diagnosed early to start treatment quickly and to avoid unnecessary morbidity. The later course seems to be more favorable than in many other intoxication-type metabolic diseases; most patients are stable and develop normally, with only a sick-day regimen. Research is ongoing to develop a reliable dried blood spot test so that this condition may be included in newborn screening panels.
Disclosure
The authors declare no conflict of interest.
Change history
15 June 2016
Carmen Diez-Fernandez PhD, Véronique Rüfenacht MSc,Saikat Santra MD, Allan M. Lund MD, DMSc, René Santer MD, Martin Lindner MD, Trine Tangeraas PhD, Caroline Unsinn MD, Pascale de Lonlay MD, Alberto Burlina MD, Clara D. M. van Karnebeek MD, PhD & Johannes Häberle MD Genet Med (2016) doi:10.1038/gim.
References
Tashian RE. The carbonic anhydrases: widening perspectives on their evolution, expression and function. Bioessays 1989;10:186–192.
Sly WS, Hu PY. Human carbonic anhydrases and carbonic anhydrase deficiencies. Annu Rev Biochem 1995;64:375–401.
Shah GN, Hewett-Emmett D, Grubb JH, et al. Mitochondrial carbonic anhydrase CA VB: differences in tissue distribution and pattern of evolution from those of CA VA suggest distinct physiological roles. Proc Natl Acad Sci USA 2000;97:1677–1682.
Nagao Y, Batanian JR, Clemente MF, Sly WS. Genomic organization of the human gene (CA5) and pseudogene for mitochondrial carbonic anhydrase V and their localization to chromosomes 16q and 16p. Genomics 1995;28:477–484.
Nagao Y, Platero JS, Waheed A, Sly WS. Human mitochondrial carbonic anhydrase: cDNA cloning, expression, subcellular localization, and mapping to chromosome 16. Proc Natl Acad Sci USA 1993;90:7623–7627.
Fujikawa-Adachi K, Nishimori I, Taguchi T, Onishi S. Human mitochondrial carbonic anhydrase VB. cDNA cloning, mRNA expression, subcellular localization, and mapping to chromosome x. J Biol Chem 1999;274:21228–21233.
van Karnebeek C, Häberle J. Carbonic anhydrase VA deficiency. In: Pagon RA, Adam MP, Ardinger HH, et al. (eds). GeneReviews®. 2015/04/04 edn. University of Washington–Seattle: Seattle, WA, 2015.
van Karnebeek CD, Sly WS, Ross CJ, et al. Mitochondrial carbonic anhydrase VA deficiency resulting from CA5A alterations presents with hyperammonemia in early childhood. Am J Hum Genet 2014;94:453–461.
Guibaud P, Larbre F, Freycon MT, Genoud J. [Osteopetrosis and renal tubular acidosis. 2 cases of this association in a sibship]. Arch Fr Pediatr 1972;29:269–286.
Vainsel M, Fondu P, Cadranel S, Rocmans C, Gepts W. Osteopetrosis associated with proximal and distal tubular acidosis. Acta Paediatr Scand 1972;61:429–434.
McMahon C, Will A, Hu P, Shah GN, Sly WS, Smith OP. Bone marrow transplantation corrects osteopetrosis in the carbonic anhydrase II deficiency syndrome. Blood 2001;97:1947–1950.
Ohlsson A, Cumming WA, Paul A, Sly WS. Carbonic anhydrase II deficiency syndrome: recessive osteopetrosis with renal tubular acidosis and cerebral calcification. Pediatrics 1986;77:371–381.
Kendall AG, Tashian RE. Erythrocyte carbonic anhydrase I: inherited deficiency in humans. Science 1977;197:471–472.
Tashian RE, Goodman M, Headings VE, Ward RH, DeSimone J. Genetic variation and evolution in the red cell carbonic anhydrase isozymes of macaque monkeys. Biochem Genet 1971;5:183–200.
DeSimone J, Magid E, Tashian RE. Genetic variation in the carbonic anhydrase isozymes of macaque monkeys. II. Inheritance of red cell carbonic anhydrase levels in different carbonic anhydrase I genotypes of the pig-tailed macaque, Macaca nemestrina. Biochem Genet 1973;8:165–174.
Lewis SE, Erickson RP, Barnett LB, Venta PJ, Tashian RE. N-ethyl-N-nitrosourea-induced null mutation at the mouse Car-2 locus: an animal model for human carbonic anhydrase II deficiency syndrome. Proc Natl Acad Sci USA 1988;85:1962–1966.
Lai LW, Chan DM, Erickson RP, Hsu SJ, Lien YH. Correction of renal tubular acidosis in carbonic anhydrase II-deficient mice with gene therapy. J Clin Invest 1998;101:1320–1325.
Shah GN, Rubbelke TS, Hendin J, et al. Targeted mutagenesis of mitochondrial carbonic anhydrases VA and VB implicates both enzymes in ammonia detoxification and glucose metabolism. Proc Natl Acad Sci USA 2013;110:7423–7428.
Alterio V, Hilvo M, Di Fiore A, et al. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc Natl Acad Sci USA 2009;106:16233–16238.
Hilvo M, Salzano AM, Innocenti A, et al. Cloning, expression, post-translational modifications and inhibition studies on the latest mammalian carbonic anhydrase isoform, CA XV. J Med Chem 2009;52:646–654.
Díez-Fernández C, Hu L, Cervera J, Häberle J, Rubio V. Understanding carbamoyl phosphate synthetase (CPS1) deficiency by using the recombinantly purified human enzyme: effects of CPS1 mutations that concentrate in a central domain of unknown function. Mol Genet Metab 2014;112:123–132.
Diez-Fernandez C, Martínez AI, Pekkala S, et al. Molecular characterization of carbamoyl-phosphate synthetase (CPS1) deficiency using human recombinant CPS1 as a key tool. Hum Mutat 2013;34:1149–1159.
Hu L, Diez-Fernandez C, Rüfenacht V, et al. Recurrence of carbamoyl phosphate synthetase 1 (CPS1) deficiency in Turkish patients: characterization of a founder mutation by use of recombinant CPS1 from insect cells expression. Mol Genet Metab 2014;113:267–273.
Häberle J, Schmidt E, Pauli S, et al. Mutation analysis in patients with N-acetylglutamate synthase deficiency. Hum Mutat 2003;21:593–597.
Kretz R, Hu L, Wettstein V, Leiteritz D, Häberle J. Phytohemagglutinin stimulation of lymphocytes improves mutation analysis of carbamoylphosphate synthetase 1. Mol Genet Metab 2012;106:375–378.
Rapp B, Häberle J, Linnebank M, et al. Genetic analysis of carbamoylphosphate synthetase I and ornithine transcarbamylase deficiency using fibroblasts. Eur J Pediatr 2001;160:283–287.
PolyPhen-2. http://genetics.bwh.harvard.edu/pph2.
MutPred. http://mutpred.mutdb.org.
Maren TH. A simplified micromethod for the determination of carbonic anhydrase and its inhibitors. J Pharmacol Exp Ther 1960;130:26–29.
Schell T, Kulozik AE, Hentze MW. Integration of splicing, transport and translation to achieve mRNA quality control by the nonsense-mediated decay pathway. Genome Biol 2002;3:REVIEWS1006.
Jank JM, Maier EM, Reiβ DD, et al. The domain-specific and temperature-dependent protein misfolding phenotype of variant medium-chain acyl-CoA dehydrogenase. PLoS One 2014;9:e93852.
Summar ML, Koelker S, Freedenberg D, et al.; European Registry and Network for Intoxication Type Metabolic Diseases (E-IMD); Members of the Urea Cycle Disorders Consortium (UCDC). The incidence of urea cycle disorders. Mol Genet Metab 2013;110:179–180.
Dercksen M, IJlst L, Duran M, et al. Inhibition of N-acetylglutamate synthase by various monocarboxylic and dicarboxylic short-chain coenzyme A esters and the production of alternative glutamate esters. Biochim Biophys Acta 2014;1842:2510–2516.
Acknowledgements
This work is supported by the Swiss National Science Foundation (grant 310030_153196). CvK’s salary is partly funded by the Michael Smith Foundation for Health Research Scholar Award. The authors acknowledge the patients, families, and referring physicians involved. The authors also thank Orphan Europe Recordati for supporting the NAGS and CPS1 mutation analysis (without influencing the planning or performance of this study or the writing of the manuscript). The Department of Medical Biochemistry, Oslo University Hospital Rikshospitalet, is acknowledged for analyzing blood and urine samples in patient 14. Finally, the authors thank Chris Ottolenghi (Paris) for his contribution to biochemical data for patient 12 and Raoul Rosenthal and Tobias Erb (both Zurich) for fruitful discussions of the CAVA enzyme assay.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Information
(DOC 110 kb)
Rights and permissions
About this article

Cite this article
Diez-Fernandez, C., Rüfenacht, V., Santra, S. et al. Defective hepatic bicarbonate production due to carbonic anhydrase VA deficiency leads to early-onset life-threatening metabolic crisis. Genet Med 18, 991–1000 (2016). https://doi.org/10.1038/gim.2015.201
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2015.201
Keywords
This article is cited by
-
Tip60 might be a candidate for the acetylation of hepatic carbonic anhydrase I and III in mice
Molecular Biology Reports (2021)
-
Presentation and management of N-acetylglutamate synthase deficiency: a review of the literature
Orphanet Journal of Rare Diseases (2020)
-
Response to Baertling et al.
Genetics in Medicine (2020)
