
Abstract
Purpose:
Genetic etiology of congenital/infantile nystagmus remains largely unknown. This study aimed to identify genomic mutations in patients with infantile nystagmus and an associated disease network.
Methods:
Patients with inherited and sporadic infantile nystagmus were recruited for whole-exome and Sanger sequencing. β-Mannosidase activities were measured. Gene expression, protein–protein interaction, and nystagmus-associated lysosomal storage disease (LSD) genes were analyzed.
Results:
A novel heterozygous mutation (c.2013G>A; p.R638H) of MANBA, which encodes lysosomal β-mannosidase, was identified in patients with autosomal-dominant nystagmus. An additional mutation (c.2346T>A; p.L749H) in MANBA was found by screening patients with sporadic nystagmus. MANBA was expressed in the pretectal nucleus of the developing midbrain, known to be involved in oculomotor and optokinetic nystagmus. Functional validation of these mutations demonstrated a significant decrease of β-mannosidase activities in the patients as well as in mutant-transfected HEK293T cells. Further analysis revealed that nystagmus is present in at least 24 different LSDs involving the brain.
Conclusion:
This is the first identification of MANBA mutations in patients with autosomal-dominant nystagmus, suggesting a new clinical entity. Because β-mannosidase activities are required for development of the oculomotor nervous system, our findings shed new light on the role of LSD-associated genes in the pathogenesis of infantile nystagmus.
Genet Med 17 12, 971–979.
Similar content being viewed by others

Introduction
Congenital/infantile nystagmus is an oculomotor disorder characterized by uncontrollable bilateral ocular oscillations. This disorder is also referred to as “idiopathic,” given that no known nervous or ocular diseases have yet to be found.1 In fact, approximately half of cases of infantile nystagmus are accompanied by structural or electrophysiological derangements involving central foveal vision in both eyes.2 For example, foveal hypoplasia and presenile cataract syndrome (OMIM 136520) or oculocutaneous albinism (OMIM 203100), which are caused by mutations in the PAX6 or the SLC38A8 gene, respectively, may be accompanied by decreased visual acuity and secondary nystagmus.
In a survey regarding genetic eye diseases among 27,193 Chinese, the prevalence of infantile nystagmus was estimated to be 0.025%,3 whereas it is 0.24% in Western countries.2 The most common cause of congenital nystagmus seems to be mutations of the X-linked FRMD7 gene (NYS1), which also have been detected by us and others in China.4 Mutations in the GPR143 gene have been found to be responsible for another X-linked ocular albinism and nystagmus (NYS6). Moreover, at least six susceptibility loci have been mapped from 1996 to 2014, including NYS2 (6p12; OMIM 164100), NYS3 (7p11; OMIM 608345), NYS4 (13q; OMIM 193003), NYS5 (Xp11.4; OMIM 300589), NYS7 (1q31-q32; OMIM 614826), and one additional locus in the X chromosome identified by a recent study.5 However, the responsible genes in the mapped loci have not been identified.
The genetic etiology and pathogenesis of infantile nystagmus remain largely unknown. Several recent hypotheses have been proposed to explain how sensory deficits could lead to nystagmus generation. One key area of controversy, however, is whether infantile nystagmus subtypes share a common mechanism. Identification of new genes and determination of their cellular functions will therefore advance our understanding of infantile nystagmus.1
Exome sequencing is a transformative technology that produces unbiased genetic data on a whole-genome scale.6 Identification of disease-causing mutations will provide a basis for determining the molecular pathogenesis of the disease and for the development of effective therapies. To identify potential genetic etiology for these ocular disorders in our hereditary disease program,4 we applied whole-exome sequencing (WES) and bioinformatics to recently recruited patients with nystagmus. In this study we report both the identification of heterozygous mutations in the MANBA gene in patients with infantile nystagmus and the molecular association of nystagmus with lysosomal storage disease (LSDs).
Materials and Methods
Patient ethics statement
Informed consent was obtained from each of the patients and, in the case of minors, from their parents. This study and associated research protocols were approved by the ethics committees of the Changzhi Medical College as well as Wenzhou Medical University.
WES and Sanger sequencing
The steps of WES and pathogenesis analysis are shown in Figure 1 . Genomic DNAs were isolated from peripheral blood leukocytes. Whole-exome capture using the SureSelect Human All Exon Kit (Agilent, Santa Clara, CA) and high-throughput sequencing by the HiSeq2000 sequencer (Illumina, San Diego, CA) were conducted in house, as previously described.7 The reads were aligned to hg19 and variants identified through the GATK pipeline. All variants were prioritized by allele frequency, conservation, and predicted effect on protein function, and they were tested for segregation with disease. Detected variants—if present in the dbSNP (www.ncbi.nlm.nih.gov/projects/SNP/), HapMap, 1000 Genomes Project (http://www.1000genomes.org/home), and ESP6500 (evs.gs.washington.edu/EVS/), and our in-house Chinese Exome Database (1,300 individuals of Chinese Han nationality)—were all removed. Deleterious single-nucleotide variants (SNVs) were predicted by SIFT (http://sift.bii.a-star.edu.sg/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), and MutationTaster (http://www.mutationtaster.org/) programs. Candidate SNVs were validated by the ABI3730 sequencer. Thirty patients with sporadic, idiopathic infantile nystagmus were screened for potential mutation evaluation of the MANBA gene by polymerase chain reaction and Sanger sequencing. Exons 1 through 17 of MANBA, including intron–exon junctions, were individually amplified by primers (Supplementary Table S2 online) and bidirectionally sequenced. The 5,499 SNVs and 172 insertions/deletions shared by three patients and normal family members (Supplementary Tables S3 and S4 online), 96 SNVs and 3 insertions/deletions presented in only three patients (Supplementary Tables S5 and S6 online), and 12 novel SNVs and 1 novel insertion/deletion in three patients (Supplementary Tables S7 and S8 online) are supplemented.

Whole-exome sequencing (WES) and pathogenesis analysis. The schematic illustrates the main steps of WES analysis and gene expression/function studies. Known or nondeleterious single-nucleotide polymorphisms and insertions/deletions were excluded. Causative genes for nystagmus and their potential interactive network are explored by Ingenuity Pathway Analysis. Detailed information of the shared single-nucleotide variants/insertions and deletions detected in the patients is provided in Supplementary Tables S3–S8 online.
Expression of MANBA
The expression data of MANBA in prenatal and adult human brains were extracted from the Allen Brain Atlas (http://www.brain-map.org/). Images of in situ hybridization for Manba transcriptional expression in mouse embryonic tissues were analyzed and selected from EURExpress and GenePaint (http://www.genepaint.org/Frameset.html). Expression levels of MANBA in the optic nerve were assessed in Gene Expression Omnibus profiles (http://www.ncbi.nlm.nih.gov/geoprofiles).
Mutagenesis and western blots
Wide-type MANBA complementary DNA (Open Biosystems, Pittsburgh, PA) was mutagenized (Mutagenesis kit; Promega, Madison, WI) to create the c.2013G>A or c.2346T>A mutation in the pcDNA3.1 vector using specific primers (Supplementary Table S2 online). HEK293T cells were then transfected with 2 μg of wild-type MANBA or R638H-MANBA or L749H-MANBA construct DNA by Lipofectamine 2000 (Invitrogen, Grand Island, NY). Cells were harvested after 48 h of culturing for enzyme activity analyses. Western blots were performed using the ECL Western Blotting System (GE Healthcare Life Sciences, Pittsburgh, PA) and MANBA mouse polyclonal antibody (1:1,000; Abnova, Taipei, Taiwan).
Enzyme assays
β-Mannosidase activity in peripheral blood leukocytes and HEK293T cells was assessed using the human β-mannosidase enzyme-linked immunosorbent assay kit (Yanji Biotech, Shanghai, China). Briefly, leukocytes or HEK293T cells were homogenized by sonication, and protein concentrations were measured by bicinchoninic acid protein assay (Pierce, Rockford, IL). Following modifications of a previous protocol,8 10 μl of the cell lysates (2 μg protein) were incubated with 20 μl of 2.0 mmol/l substrate 4-methylumbelliferyl-β-D-mannopyranoside (4MU-β-mannoside; Sigma, St. Louis, MO) in a reaction buffer (0.2 M sodium phosphate/0.1 M citric acid buffer; pH 4.2) at 37 °C for 1 h. After the reaction was stopped with 200 μl of the buffer (0.5 M Na2CO3/NaHCO3; pH 10.7), the fluorescence of 4-MU was measured (excitation, 362 nm; emission, 448 nm) using a Multi-label Counter 1420 Microplate Reader (Perkin Elmer, Waltham, MA). All experiments were performed independently at least three times, and each experiment consisted of triplicate samples.
Nystagmus in other inherited LSDs
A list of genes that share biological similarity to MANBA was generated through careful curation of OMIM (http://www.ncbi.nlm.nih.gov/omim/) gene entries implicated as causative in LSDs involving the brain. Convergent pathogenic pathways were predicted and assessed by protein–protein interaction network (Ingenuity Pathway Analysis; http://www.ingenuity.com/).
Results
Clinical presentations
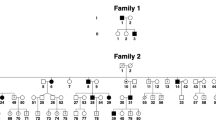
Three patients with idiopathic infantile nystagmus and reduced visual acuity (i.e., bilateral correctable low vision) in a nonconsanguineous family (HH02) were recruited at Heping Hospital in Northwestern China ( Figure 2a ). The diagnosis was made based on the 10 general features of infantile nystagmus.9 The patients showed horizontal, conjugate, and pendular nystagmus predominant in the primary position ( Table 1 ) since 2–3 months of age. No colorblindness, oscillopsia, vertigo, or imbalance were found. No angiokeratomas, organomegaly, coarse facial features, or dysostosis multiplex were found. No other abnormalities were found during a general physical examination. No other neurologic abnormalities outside of the visual system were identified. Optical coherence tomography, flash electroretinography, visual evoked potential examinations, and brain magnetic resonance imaging did not show any noticeable abnormality in the patients (Supplementary Figure S3 online).

MANBA mutations and β-mannosidase structure. (a) Pedigree of the family. (b) The c.2013G>A mutation of MANBA is confirmed in all patients and the carrier (I-2) by Sanger sequencing. (c) An additional patient (ZB005) with infantile nystagmus. (d) The c.2346A>T mutation of MANBA is identified. Filled squares or circles represent subjects with nystagmus. The arrows denote the probands. (e) The encoded protein with two enzymatic active domains is a member of the glycosyl hydrolase 2 family. Multiple sequence alignment shows the evolutionary conservation of the indicated residues. The β-mannosidosis disorder caused by MANBA mutations has been identified in the four species listed. (f) The residue R638 (in red) and L749 (in orange) were positioned in the three-dimensional structural model. (g) The residue R638 (in red) and R641 (in yellow) are predicted to be associated with D709 (in blue).
To identify potential causative mutations, we performed WES in seven members of the family, including three patients with autosomal-dominant nystagmus and four unaffected subjects. An average of 57.82 Mb of aligned base reads per sample were generated, of which 96.93% of reads reached ≥10× coverage (Supplementary Table S1 online). The pathogenic cause was assumed to be the same heterozygous mutation in a single gene. A total of 39,333 SNVs (missense, nonsense, and splice-site mutations) and 1,480 insertions/deletions (short coding insertions or deletions) were identified in the three patients ( Figure 1 ). After the known variants (present in the normal family members or in the databases as mentioned in the Materials and Methods) and nondeleterious SNVs were removed, the candidate genes were reduced to five. After bioinformatic analyses, we excluded four unrelated genes (DOK3, ASB16, FGD6, and PRDM2) because variants of these genes were nonpathogenic based on SIFT/PolyPhen-2/MutationTaster analyses and because the functions of these genes are unrelated to the visual system. Finally, the MANBA gene was prioritized because mutations in the gene have caused a wide spectrum of neurological disorders in both humans and animals. More convincingly, nystagmus also has been observed in ruminants such as goats and cattle with β-mannosidosis10 caused by disruptive homozygous mutations of MANBA.11
Identification of mutations
Using bidirectional Sanger sequencing with specific primers (Supplementary Table S2 online), we confirmed the novel inherited heterozygous mutation (c.2013G>A) in exon 14 of the MANBA gene at chromosome 4q25 (GenBank accession no. NM_005908.3) in all three patients (Sanger sequencing data of proband III-2 are shown in Figure 2b ), but not in the unaffected family members I-3, II-2, and II-3 (data not shown). This mutation changed the codon 638 CGC for arginine into histidine (p.R638H), resulting in a missense mutation. This mutation is segregated with the disease phenotype and predicted to be deleterious or to cause disease by three different programs: SIFT (affecting protein function, with a score of 0.01), PolyPhen-2 (probably damaging, with a score of 0.977), and MutationTaster (disease causing, with a score of 0.983). Also, this mutation was not found in the ESP6500, 1000 Genomes Project, or the in-house exome databases, suggesting that it is a private SNV cosegregated with the phenotype nystagmus in the family. Subject I-2 was the obligate carrier because of reduced penetrance. Reduced penetrance also was observed in the FRMD7-associated nystagmus, probably as a result of the type of mutation in FRMD7 and random inactivation of the X chromosome.12 Furthermore, haplotype analysis with single-nucleotide polymorphisms flanking the MANBA locus confirmed that the c.2013G>A mutation-containing region segregates with the patients’ phenotypes and also matches the haplotype of the obligate carrier (Supplementary Table S10 online). In addition, with the exception of decreased vision, no other medical history was documented, and whether her sister (I-1) had nystagmus or not is unknown.
To examine whether additional mutations in MANBA could be identified in unrelated patients, we used polymerase chain reaction and Sanger sequencing to screen the whole coding region of the gene, including 17 exons and intron–exon junctions (Supplementary Table S2 online), in a cohort of 30 patients with sporadic, idiopathic infantile nystagmus. As a result, we found one more heterozygous missense mutation (c.2346T>A, p.L749H) in patient ZB005 ( Figure 2c , d ). This substitution was not shown in the in-house Chinese exome database (see Methods), and it was predicted to be harmful by the three programs mentioned above. Because the parent DNAs were unavailable, whether c.2346T>A is an inherited or de novo mutation was not determined.
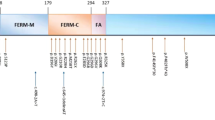
To illustrate the potential effects of the mutations on the protein’s function, the locations of p.R638H and p.L749H in β-mannosidase were compared with four known missense mutations that cause recessive β-mannosidosis, which is one LSD ( Figure 2e ). Multiple sequence alignment of a region of human MANBA harboring the mutations with that of goats, cattle, and cats showed that both p.R638 and p.L749 are evolutionarily conserved in the four species, and β-mannosidosis has been previously reported in these species ( Figure 2e , lower panel; also see Supplementary Figure S4 online).
Furthermore, structural bioinformatics analysis showed that the last domain from residue 572 to 879 ( Figure 2e , f ) stabilizes the β-mannosidase active sites (residues 342–571).13 Because the positively charged arginine 638 was substituted by a slightly positive histidine, p.R638H may destabilize β-mannosidase by affecting the salt bridge D709 or the interaction between R641 and D709, as shown in a three-dimensional structural model ( Figure 2f , g ) that was established using PyMOL (L. Schrödinger, version 1.3r1; PDB ID code 2JE8). This assumption is supported by previous analysis of the p.R641H mutation, which caused β-mannosidosis deficiency.14
Analysis of MANBA localization in brain tissues
To examine whether MANBA is expressed in the brain region involving regulation of eye movement, we compared publicly accessible transcriptional profiles. Expression analysis of the Allen Brain Atlas showed that MANBA was localized in various brain regions, primarily restricted to the cerebellar cortex, midbrain, pons, thalamus, and corpus callosum (Supplementary Table S9 online). MANBA also was expressed at a low level across all periods of human brain development, from 8 weeks after conception to adult stages (Supplementary Figure S1 online). Furthermore, an in situ hybridization study of whole-mount mouse embryos at embryonic day 14.5 showed that MANBA was expressed in the central nervous system (CNS), particularly in the pretectum of the midbrain ( Figure 3a – c ), which primarily involves oculomotor and optokinetic nystagmus.15 MANBA also was abundantly expressed in the optic nerve of humans (National Center for Biotechnology Information, Gene Expression Omnibus profile ID no. 4031643) and mouse (Gene Expression Omnibus profile ID no. 71898221). Notably, light and electron microscopic analyses showed a substantial decrease in the number of myelinated axons in the fetal optic nerve of β-mannosidosis-affected goats.16 Overall, the expression of MANBA was relatively restricted to the mid- and hindbrain (i.e., the cerebellum, pons, and medulla), regions known to be involved in the optokinetic nystagmus neural pathway. These findings also revealed that in β-mannosidosis, pathological alterations in the optic nerve and pretectal region are involved in the oculomotor pathway of the brain ( Figure 3d ).

MANBA expression and β-mannosidase activity. (a) Sagittal in situ hybridization image in a mouse embryo at embryonic day 14.5 (GenePaint) shows Manba expression in the pretectum of the midbrain. (b) A similar pattern is also shown using EURExpress. (c) Threefold amplification of the midbrain. (d) Dorsal view of the afferent and efferent pathways. The impulse starts in the retina and crosses over the optic chiasm to synapse in the pretectal nucleus, and then crosses over to synapse in the nucleus of cranial nerve III, which is involved in ocular movement. Regions with pathological alterations in animals with β-mannosidosis are indicated. (e) β-Mannosidase activity in patients and controls. (f) MANBA protein level is shown by western blots. A mock, nontransfected control was used. (g) β-Mannosidase activity in HEK293 cells (mean ± SEM from three independent transfections conducted in triplicate). *P < 0.05; **P < 0.01.
Reduced β-mannosidase activity
To examine whether heterozygous mutations affect the function of MANBA-encoded protein, we measured β-mannosidase activity in peripheral blood leukocytes collected from 5 available family members as well as 20 unrelated normal controls. Using a human β-mannosidase enzyme-linked immunosorbent assay kit, we found a 40–60% reduction of β-mannosidase activity in the patients (II-1 and III-2) and carrier (I-2) compared with the normal family members (I-3, II-3; Figure 3e ) and the 20 unrelated normal controls. This result indicates that the subjects with heterozygous mutations only have intermediate levels of normal β-mannosidase activity in peripheral blood leukocytes (~35 nmol/h.mg of protein). To test whether the c.2013G>A and c.2346T>A mutations are responsible for the reduced β-mannosidase activity in the patients, we expressed the wild-type and mutated MANBA genes in HEK293T cells. As shown in the western blots, the levels of mutant β-mannosidase proteins (R638H and L749H) were slightly less abundant than the wild-type MANBA ( Figure 3f ), suggesting possible instability of the mutants. As expected, the β-mannosidase activity in the mutant-transfected cells was much lower than that in the wild-type MANBA-transfected cells ( Figure 3g ), indicating that the mutant β-mannosidase produced by either R638H- or L749H-containing construct exhibited only very low enzymatic activity. In addition, by using a lysosomal enrichment kit and western blot analysis, we confirmed that these β-mannosidase proteins were still located in the lysosome (data not shown).
Nystagmus and LSDs
Because nystagmus is genetically heterogeneous, protein–protein interaction and associated disease network analyses may provide the landscape of nystagmus etiology. The association between the lysosomal gene MANBA and nystagmus prompted us to examine whether nystagmus is also present in other LSDs. After an extensive evaluation of OMIM entries of LSDs involving the brain, we curated a total of 27 LSDs involving brain, which can be divided into five categories ( Table 2 ). Surprisingly, by genotype–phenotype analysis of each of the diseases, we found that nystagmus was shown in most, if not all, LSDs involving the CNS.17 In particular, patients with ceroid lipofuscinosis CLN4B caused by heterozygous mutations of DNAJC5 have also been reported with nystagmus in the autosomal-dominant form, like our case.18
To find potentially shared pathophysiological mechanisms at the molecular and cellular levels, we performed Ingenuity Pathway Analysis for the causative genes of these 27 LSDs. As a result, these genes are implicated in multiple lysosomal functions, such as neuronal development, lipid metabolism, molecular transport, small-molecule biochemistry, cellular assembly and organization, and cellular function and maintenance. As an example of these complex interactions, a network of protein–protein interactions that depict an interconnection of the 16 lysosome-associated genes involving neuronal development and nystagmus is illustrated in Supplementary Figure S2 online.
Discussion
Using WES analysis, we identified a novel MANBA mutation in patients with inherited nystagmus. To the best of our knowledge, this is the first identification of infantile nystagmus in an autosomal-dominant form linked to a causative gene of inherited disorders. The following provide evidence for a pathogenic role of MANBA mutations in nystagmus: (i) MANBA is the only candidate deleterious gene linked to the nystagmus phenotype of all affected subjects. (ii) An additional rare deleterious mutation was identified by screening a cohort of patients with the same disorder. (iii) MANBA is expressed in brain regions involving eye movements and nystagmus; the expression pattern of MANBA in the brain is also similar to the pattern of the X-linked nystagmus gene FRMD7 in the developing brain.19 (iv) Functional assays demonstrated that β-mannosidase activity in the samples from affected subjects is significantly decreased. (v) In particular, nystagmus and substantially reduced myelinated axons in the fetal optic nerve have been observed in ruminant animals with MANBA mutations. (vi) Gene–disease pathway analysis has revealed that a majority of LSDs involving the brain are accompanied by the nystagmus phenotype.
Normal functioning of the lysosome is important for degradation of macromolecules and cell homeostasis. The lysosome-endosomal system is intimately involved in the regulation of autophagy, apoptosis, and cell death involving in neurological diseases and many other conditions.20 LSDs are a group of rare, hereditary metabolic disorders that result from mutations in genes encoding catabolic enzymes for the degradation of macromolecules. The accumulated substrate(s) of the defective enzyme(s) lead to dysfunction in the nervous system, eye, and other systems and ultimately lead to cell death and organ-specific clinical manifestations. Most LSDs are inherited in an autosomal-recessive manner,21 but some, like patients with neuronal ceroid lipofuscinosis 4B and nystagmus18,22 and the case presented here, present autosomal-dominant inheritance.23,24
Mutations in MANBA have been previously linked to β-mannosidosis (OMIM 248510), an autosomal-recessive LSD resulting in demyelination of the CNS. The encoded β-mannosidase subcellularly localizes to the lysosome, where it is the final exoglycosidase for N-linked glycoprotein oligosaccharide catabolism. Patients with β-mannosidosis have a wide spectrum of neurological involvement, including intellectual disability, hearing loss, ataxia, and seizures.13 Phenotype–genotype correlation analysis revealed that truncation mutations often cause severe phenotypes, whereas substitution mutations may show mild phenotypes (Supplementary Table S11 online).13
This study extends the scope of β-mannosidosis to the autosomal-dominant inheritance of infantile nystagmus. The heterozygous mutations in MANBA identified in this study may represent the least severe β-mannosidosis that is limited to the eye regulatory regions in brain. Because of significantly reduced β-mannosidase activity in these patients ( Figure 3 ), the substrate N-linked glycoprotein oligosaccharide is expected to build up over time, leading to excess cellular storage of this material and potentially leading to neuronal cell death and hypomyelination.
Nystagmus has been identified as one of the main signs of demyelinating or hypomyelinating leukoencephalopathies in the CNS of ruminants with β-mannosidosis.10,25,26,27 In particular, microscopic analysis observed a substantial decrease in the number of myelinated axons and a decreased proportion of oligodendrocytes in the fetal optic nerve of β-mannosidosis-affected goats,16 indicating that the pathogenetic process was initiated during an early stage of myelination in the optic nerve ( Figure 3d ). Another study confirmed that oligosaccharide accumulates in the regional CNS in caprine β-mannosidosis.28 These findings clearly link the prenatal lesions of the optic nerve and regional CNS in β-mannosidosis to ocular motor phenotypes such as infantile nystagmus in goats. Genetic inactivation of the MANBA gene in mice, however, did not show signs of lysosomal storage in neuronal tissues or β-mannosidosis-related phenotypes,29 suggesting a compensatory mechanism that might be specific to the mouse.
As a matter of fact, demyelination occurs in many other LSDs in brain. Because each neuronal cell or glial cell subtype can synthesize different sphingolipids, LSD pathologies can be cell specific, and different disorders can manifest with diverse neurological syndromes.30 Abnormal eye movements can present as initial clinic signs in type 2 Gaucher disease, as a major sign in type 3 Gaucher disease, and as an early sign in many other LSDs17 ( Table 2 ). Also, distinct eyemovement disorders occurred in late-onset GM2-gangliosidosis disease (i.e., Tay-Sachs disease), Gaucher disease type 3, and Niemann–Pick type C. For instance, limitation of the upward gaze is a common finding in Niemann–Pick type C and serves as a valuable clinical sign of this disorder. Optic nerve atrophy and retinopathy are common in neuronal ceroid lipofuscinoses and some classic LSDs that profoundly affect the brain and optic nerves, causing blindness and eyemovement disorders.30 The shared clinical features suggest that the various proteins operate in similar functional pathways in these disorders.
Interestingly, the most common cause of acquired nystagmus probably is multiple sclerosis,31 in which the nerves are demyelinated in the brain and spinal cord. Notably, MANBA (the molecular marker rs228614) has been recently identified as a genetic risk locus for multiple sclerosis by genome-wide association study analyses of a large population,32 suggesting that this polymorphic allele of MANBA might be associated with nystagmus in multiple sclerosis.
Nystagmus may also be observed in non-LSDs in which myelin synthesis is impaired. As an example, nystagmus frequently occurs in microcephaly disorder resulting from mutations of CASK.33,34 Yet, the G-protein-coupled receptor 143 encoded by GPR143, which is responsible for X-linked congenital nystagmus 6, also targets the lysosome35 and delays delivery of multivesicular bodies, not the GPR143 gene product itself, to the lysosome.36 Many G-protein-coupled receptors regulate myelin-associated glycoprotein in neuronal cells. Moreover, it is worth noting that nystagmus has been proposed as a key clinical symptom for the diagnosis of CNS hypomyelination.37 Nystagmus as an early ocular alteration was recently present in 88% of patients with autosomal-dominant Machado–Joseph disease and 17% of carriers who were nonpenetrant for the disease, but it did not occur in those who did not carry the mutation.38 These observations are in agreement with the hypothesis that impairment of neuronal connections between subcortical and cortical optokinetic pathways may be associated with pathogenesis of congenital/infantile nystagmus.9
Nevertheless, how the mutations in MANBA and other lysosomal disease–associated genes are attributed to the pathogenesis of nystagmus is still not clear. Variable phenotypes and reduced penetrance, as well as potential genetic modifiers, may also contribute to the underlying complex mechanisms. Theoretically, generation and control of eye movements require the participation of the cortex, cerebellum, and brain stem.39 Accordingly, the bifurcation model has proposed that the waveform variability in the network between the brain stem and cerebellum is associated with hereditary eye-movement disorders.40 Our findings presented here seem to support this hypothesis in terms of the association of this network with abnormal connectivity or impaired plasticity of neurons in these tissues.
Given the fact that nystagmus is often described as an accompanying symptom in many different disorders, including LSDs, our findings instead indicate that nystagmus may present as an isolated or early clinical manifestation in these disorders as a result of subtle mutations or compensatory mechanisms. Therefore, developing gene-specific enrichment methods for molecular examination of undiagnosed nystagmus is conceivable. In the case of LSDs, earlier identification of mutations may raise questions as to whether patients should receive enzyme replacement therapy; this will require further study.
Taken together, our results provide compelling evidence that the heterozygous mutations of MANBA identified here are responsible for infantile nystagmus. This is the first identification of an autosomal-dominant nystagmus, suggesting a new clinical entity in LSDs involving the brain. Furthermore, gene–disease network analyses suggest that mutations in LSD-related genes, as well as other causative genes, which affect myelination and development of oculomotor nervous system, may play an important role in the pathogenesis of eye-movement disorders.
Disclosure
The authors declare no conflict of interest.
References
Gottlob I, Proudlock FA. Aetiology of infantile nystagmus. Curr Opin Neurol 2014;27:83–91.
Sarvananthan N, Surendran M, Roberts EO, et al. The prevalence of nystagmus: the Leicestershire nystagmus survey. Invest Ophthalmol Vis Sci 2009;50:5201–5206.
Hu DN. Prevalence and mode of inheritance of major genetic eye diseases in China. J Med Genet 1987;24:584–588.
Zhang X, Ge X, Yu Y, et al. Identification of three novel mutations in the FRMD7 gene for X-linked idiopathic congenital nystagmus. Sci Rep 2014;4:3745.
Deng N, Tang F, Yang Y, Chen Y, Zou W. Mapping the disease gene in two congenital motor nystagmus families. Clin Experiment Ophthalmol 2014;42:94–96.
Evans JP. Finding common ground. Genet Med 2013;15:852–853.
Wu J, Shen E, Shi D, Sun Z, Cai T. Identification of a novel Cys146X mutation of SOD1 in familial amyotrophic lateral sclerosis by whole-exome sequencing. Genet Med 2012;14:823–826.
Panday RS, van Diggelen OP, Kleijer WJ, Niermeijer MF. beta-Mannosidase in human leukocytes and fibroblasts. J Inherit Metab Dis 1984;7:155–156.
Brodsky MC, Dell’Osso LF. A unifying neurologic mechanism for infantile nystagmus. JAMA Ophthalmol 2014;132:761–768 .
Jones MZ, Cunningham JG, Dade AW, et al. Caprine beta-mannosidosis: clinical and pathological features. J Neuropathol Exp Neurol 1983;42:268–285.
Leipprandt JR, Chen H, Horvath JE, Qiao XT, Jones MZ, Friderici KH. Identification of a bovine beta-mannosidosis mutation and detection of two beta-mannosidase pseudogenes. Mamm Genome 1999;10:1137–1141.
Self J, Lotery A. A review of the molecular genetics of congenital Idiopathic Nystagmus (CIN). Ophthalmic Genet 2007;28:187–191.
Huynh T, Khan JM, Ranganathan S. A comparative structural bioinformatics analysis of inherited mutations in β-D-Mannosidase across multiple species reveals a genotype-phenotype correlation. BMC Genomics 2011;12(suppl 3):S22.
Labauge P, Renard D, Castelnovo G, Sabourdy F, de Champfleur N, Levade T. Beta-mannosidosis: a new cause of spinocerebellar ataxia. Clin Neurol Neurosurg 2009;111:109–110.
Gamlin PD. The pretectum: connections and oculomotor-related roles. Prog Brain Res 2006;151:379–405.
Lovell KL, Boyer PJ. Dysmyelinogenesis in caprine beta-mannosidosis: ultrastructural and morphometric studies in fetal optic nerve. Int J Dev Neurosci 1987;5:243–253.
Prada CE, Grabowski GA. Neuronopathic lysosomal storage diseases: clinical and pathologic findings. Dev Disabil Res Rev 2013;17:226–246.
Boehme DH, Cottrell JC, Leonberg SC, Zeman W. A dominant form of neuronal ceroid-lipofuscinosis. Brain 1971;94:745–760.
Tarpey P, Thomas S, Sarvananthan N, et al. Mutations in FRMD7, a newly identified member of the FERM family, cause X-linked idiopathic congenital nystagmus. Nat Genet 2006;38:1242–1244.
Miller JA, Ding SL, Sunkin SM, et al. Transcriptional landscape of the prenatal human brain. Nature 2014;508:199–206.
Zhong N, Moroziewicz DN, Ju W, et al. Heterogeneity of late-infantile neuronal ceroid lipofuscinosis. Genet Med 2000;2:312–318.
Verma R, Raut TP, Tiwari N, Malhotra KP, Hussain N, Malhotra HS. Late infantile neuronal ceroid lipofuscinosis: a case report with review of literature. Ann Indian Acad Neurol 2013;16:282–285.
Nijssen PC, Brusse E, Leyten AC, Martin JJ, Teepen JL, Roos RA. Autosomal dominant adult neuronal ceroid lipofuscinosis: parkinsonism due to both striatal and nigral dysfunction. Mov Disord 2002;17:482–487.
Nosková L, Stránecký V, Hartmannová H, et al. Mutations in DNAJC5, encoding cysteine-string protein alpha, cause autosomal-dominant adult-onset neuronal ceroid lipofuscinosis. Am J Hum Genet 2011;89:241–252.
Shapiro JL, Rostkowski C, Little PB, Smith-Maxie L, Julian RJ, Van Dreumel AA. Caprine beta-Mannosidosis in Kids from an Ontario Herd. Can Vet J 1985;26:155–158.
Kumar K, Jones MZ, Cunningham JG, Kelley JA, Lovell KL. Caprine beta-mannosidosis: phenotypic features. Vet Rec 1986;118:325–327.
Lovell KL. Caprine beta-mannosidosis: development of glial and myelin abnormalities in optic nerve and corpus callosum. Glia 1990;3:26–32.
Boyer PJ, Jones MZ, Rathke EJ, Truscott NK, Lovell KL. Regional central nervous system oligosaccharide storage in caprine beta-mannosidosis. J Neurochem 1990;55:660–664.
Zhu M, Lovell KL, Patterson JS, Saunders TL, Hughes ED, Friderici KH. Beta-mannosidosis mice: a model for the human lysosomal storage disease. Hum Mol Genet 2006;15:493–500.
Boustany RM. Lysosomal storage diseases–the horizon expands. Nat Rev Neurol 2013;9:583–598.
Tilikete C, Jasse L, Pelisson D, et al. Acquired pendular nystagmus in multiple sclerosis and oculopalatal tremor. Neurology 2011;76:1650–1657.
Lill CM, Schjeide BM, Graetz C, et al. MANBA, CXCR5, SOX8, RPS6KB1 and ZBTB46 are genetic risk loci for multiple sclerosis. Brain 2013;136:1778–1782.
Tarpey PS, Smith R, Pleasance E, et al. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat Genet 2009;41:535–543.
Anitei M, Ifrim M, Ewart MA, et al. A role for Sec8 in oligodendrocyte morphological differentiation. J Cell Sci 2006;119(Pt 5):807–818.
Piccirillo R, Palmisano I, Innamorati G, Bagnato P, Altimare D, Schiaffino MV. An unconventional dileucine-based motif and a novel cytosolic motif are required for the lysosomal and melanosomal targeting of OA1. J Cell Sci 2006;119(Pt 10):2003–2014.
Burgoyne T, Jolly R, Martin-Martin B, et al. Expression of OA1 limits the fusion of a subset of MVBs with lysosomes - a mechanism potentially involved in the initial biogenesis of melanosomes. J Cell Sci 2013;126(Pt 22):5143–5152.
Bassani R, Pareyson D, D’Incerti L, Di Bella D, Taroni F, Salsano E. Pendular nystagmus in hypomyelinating leukodystrophy. J Clin Neurosci 2013;20:1443–1445.
Raposo M, Vasconcelos J, Bettencourt C, Kay T, Coutinho P, Lima M. Nystagmus as an early ocular alteration in Machado-Joseph disease (MJD/SCA3). BMC Neurol 2014;14:17.
Moncayo J, Bogousslavsky J. Eye movement abnormalities. Front Neurol Neurosci 2012;30:13–16.
Barreiro AK, Bronski JC, Anastasio TJ. Bifurcation theory explains waveform variability in a congenital eye movement disorder. J Comput Neurosci 2009;26:321–329.
Acknowledgements
This work was supported by research funding from Wenzhou Medical University and Zhejiang Province Nature Science Research (to P.Y.), the National Natural Science Foundation of China and the Natural Science Foundation of Zhejiang Province (81110114 and Z2110521, respectively, to Z.S.S.), the Foundation of Shanxi Province Health Bureau (201302017 to Y.C.), and by the National Key Basic Research Program (2013CB967502 to Z.-B.J. and 2011CB504600 to J.Q.). The authors thank the patients and their families for their participation in this study as well as Jinyu Wu and Qianzhi Shao and all other co-workers involved in the WES analysis.
Author information
Authors and Affiliations
Corresponding authors
Supplementary information
Supplementary Information
(ZIP 7143 kb)
Rights and permissions
About this article

Cite this article
Yu, P., Cui, Y., Cai, W. et al. Lysosomal storage disease in the brain: mutations of the β-mannosidase gene identified in autosomal dominant nystagmus. Genet Med 17, 971–979 (2015). https://doi.org/10.1038/gim.2015.10
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2015.10
Keywords
This article is cited by
-
Three intellectual disability-associated de novo mutations in MECP2 identified by trio-WES analysis
BMC Medical Genetics (2020)
-
Identification of Novel Compound Mutations in PLA2G6-Associated Neurodegeneration Patient with Characteristic MRI Imaging
Molecular Neurobiology (2017)
