
Abstract
Purpose:
Marfan syndrome is a systemic disorder that typically involves FBN1 mutations and cardiovascular manifestations. We investigated FBN1 genotype–phenotype correlations with aortic events (aortic dissection and prophylactic aortic surgery) in patients with Marfan syndrome.
Methods:
Genotype and phenotype information from probands (n = 179) with an FBN1 pathogenic or likely pathogenic variant were assessed.
Results:
A higher frequency of truncating or splicing FBN1 variants was observed in Ghent criteria–positive patients with an aortic event (n = 34) as compared with all other probands (n = 145) without a reported aortic event (79 vs. 39%; P < 0.0001), as well as Ghent criteria–positive probands (n = 54) without an aortic event (79 vs. 48%; P = 0.0039). Most probands with an early aortic event had a truncating or splicing variant (100% (n = 12) and 95% (n = 21) of patients younger than 30 and 40 years old, respectively). Aortic events occurred at a younger median age in patients with truncating/splicing variants (29 years) as compared with those with missense variants (51 years). A trend toward a higher frequency of truncating/splicing variants in patients with aortic dissection (n = 21) versus prophylactic surgery (n = 13) (85.7 vs. 69.3%; not significant) was observed.
Conclusion:
These aortic event– and age-associated findings may have important implications for the management of Marfan syndrome patients with FBN1 truncating and splicing variants.
Genet Med 17 3, 177–187.
Similar content being viewed by others


Introduction
FBN1 mutations are most commonly associated with Marfan syndrome (MFS), an autosomal dominant connective tissue disorder typically involving the ocular, skeletal, and cardiovascular systems; MFS less frequently involves the skin, integument, lung, muscle, and adipose tissue. Cardiovascular manifestations, which are the major cause of morbidity and early mortality in MFS, include aortic dilatation at the level of the sinus of Valsalva, predisposition for aortic dissection, mitral valve and tricuspid valve prolapse, and enlargement of the proximal pulmonary artery.
In MFS, an age-dependent association with the occurrence of an aortic event (aortic dissection or prophylactic aortic surgery) has been demonstrated in 16% of 30–year-olds and 74% of 60-year-olds having an aortic event.1 A sex-dependent association has also been observed: aortic events occur at an earlier age in males than in females.1
Hundreds of mutations have been identified in FBN1—many of them unique to individual families. Missense mutations are the most common type of FBN1 mutation, the majority of which are cysteine substitutions. FBN1 mutations have been shown to occur across the gene with limited genotype–phenotype correlations, with the exception of the association of early onset, severe (previously termed “neonatal”) MFS and mutations in exons 24 through 32, as well as the association of ectopia lentis with missense mutations. A study investigating genotype–phenotype correlations with cardiovascular features did not observe significant differences with mutation type (e.g., frameshift versus missense) but did observe a higher probability of ascending aortic dilatation, aortic event, and mitral valve prolapse in patients with mutations altering a cysteine residue.1
The type of FBN1 mutation identified and its likelihood of being pathogenic are recognized as important factors when making a diagnosis of MFS and later clinical decisions. Some have argued that truncating and splicing mutations may be associated with a milder disease course. Accordingly, we evaluated 179 consecutive probands with FBN1 mutations to evaluate this important clinical issue.
Materials and Methods
Study population
Proband samples (n = 179) with a pathogenic or likely pathogenic FBN1 variant and detailed clinical information received over a 4.25-year period were included in this study. Each patient was examined by his or her referring physician. For Mayo Clinic patients, phenotypic information was extracted from the patients’ electronic medical record. For patients external to the Mayo Clinic, phenotypic information was provided by the referring provider via a requisition form specific to FBN1, which included age, sex, suspected diagnosis, family history, and phenotypic features, including those related to the Ghent (2010 revised) nosology criteria.2 The ethnicities of patients with reported aortic aneurysm/dissection and/or prophylactic aortic root surgery (n = 34) were European Caucasian (n = 21), African American (n = 7), Hispanic (n = 2), Arabic (n = 1), and unspecified (n = 3). The study was approved by the Mayo Foundation institutional review board.
FBN1 sequencing
Genomic DNA was extracted from ethylenediaminetetraacetic acid–anticoagulated whole blood. All 65 exons of FBN1 (RefSeq NM_000249.3) and a minimum of 20 base pairs (bp) of intronic DNA flanking each exon were amplified by multiplexed polymerase chain reaction (PCR). Amplification was performed using a common master mix containing Platinum Taq DNA Polymerase, 10× PCRx Enhancer System, 10× PCR buffer (minus magnesium chloride), magnesium sulfate (all Invitrogen, Carlsbad, CA), and a 10 mmol/l deoxynucleotide triphosphate mixture (Roche, Indianapolis, IN). Master mix and forward and reverse primers were combined with genomic DNA and amplified by 35 cycles of PCR (30 s at 95 °C; 30 s initially at 68 °C then decreased by 0.5 °C each cycle, with the last 20 cycles performed at 60 °C; and a 1-min extension at 72 °C, with a final 10-min extension at 72 °C). Amplicons were bidirectionally sequenced using Big Dye Terminator technology on an ABI 3730 system (Applied Biosystems, Foster City, CA). Sequence analysis was done using Mutation Surveyor software (SoftGenetics, State College, PA) and visual inspection.
Classification of alterations
FBN1 alteration nomenclature was based on RefSeq NM_000138.4. FBN1 variants were analyzed for pathogenicity based on criteria that included (i) whether they were previously reported and had associated functional studies; (ii) the nature of the variant (e.g., missense, nonsense); (iii) the location of the variant (e.g., critical cysteine residue of calcium-binding epidermal growth factor-like (cbEGF-like) domain); and (iv) the frequency of the variant in the Exome Variant Server database and the Single Nucleotide Polymorphism Database. Variants were classified as pathogenic if they were nonsense point mutations, frameshift insertions/deletions, or mutations involving the splice donor (intron +1G or +2T) or splice acceptor (intron −1G or −2A). Variants were classified as likely pathogenic if the variant affected or created a cysteine residue in a cbEGF-like or transforming growth factor-β binding protein domain; if it affected a known consensus/critical residue (e.g., the critical glycine at position 3 between cysteine 2 and cysteine 3 in the cbEGF-like domain); if there was a previous literature report describing a negative impact of the variant on protein function; and/or if the variant was determined to be de novo or otherwise likely pathogenic based on family studies.
Statistical analyses
Statistical analyses were performed when appropriate using Fisher’s exact test with GraphPad Software (GraphPad is a free Web-based application available at http://www.graphpad.com).
Results
A total of 179 probands with pathogenic and likely pathogenic variants fell into the following categories: 96 missense (53%), 59 (33%) nonsense or frameshift (truncating), and 24 (13%) splicing. Of these, 32 probands had a reported personal history of aortic events (aortic dissection (with or without surgical repair) or prophylactic aortic surgery), and two young Ghent-positive probands (15 and 21 years old) with aortic root dilatation had a detailed paternal family history of aortic events. All 34 of these individuals fulfilled the 2010 revised Ghent nosology criteria. Characteristics of patients with aortic events as compared with patients without a reported aortic event are described in Table 1 . Detailed genetic and clinical information about the aortic event cohort is listed in Table 2 .
The majority (79%) of the observed variants in this aortic event group were protein truncating (62%: nonsense (12/34; 35%) and frameshift (9/34; 27%)) or splicing (6/34; 18%), and only 21% (7/34) of them were missense mutations ( Table 1 ). In comparison, protein truncating or splicing variants occurred at a frequency of 39% (truncating, 27%; splicing, 12%) in mutation-positive patients without a reported aortic event. An analysis of mutation-positive patients without a reported aortic event who fulfilled the 2010 revised Ghent nosology criteria demonstrated that 52% (28/54) of these patients had a missense variant. Thus, among Ghent-positive patients, there was also a significantly lower frequency of truncating/splicing variants in those who did not have a reported aortic event (48 vs. 79%; P = 0.0039). Because our aortic event cohort did not contain patients younger than 15 years of age, and because younger patients are much less likely to have aortic events, we performed a secondary analysis of the cohort, excluding patients younger than 15 years of age. Patients without a reported aortic event who were 15 years of age or older had a similar frequency of truncating/splicing variants (41%) as patients without a reported aortic event at all ages (39%) ( Table 1 ), and this frequency was still statistically significantly lower than the 79% observed in patients with aortic events (P = 0.0003).
Détaint et al.1 previously reported on cardiovascular manifestations in 1,013 probands with pathogenic FBN1 mutations. They observed that the probability of an aortic event before the age of 30 years in their cohort was 16% in men and 11% in women; before the age of 40 years this probability was ~40–45% in men and 25% in women. Because our aortic event cohort had a wide age range (15–71 years) at the time of testing, we performed additional age-dependent analyses. There were 12 patients with aortic events (6 male and 6 female) younger than the age of 30, and all of them (12/12; 100%) had a truncating or splicing variant. When we included patients 40 years of age or younger with an aortic event, 20 of 21 patients (95%; 9 males, 12 females) had a truncating or splicing variant. The one patient in this group who did not have a truncating or splicing variant had aortic root replacement surgery at the age of 34 years (aortic root diameter, 51 mm), and she harbored a novel, likely pathogenic p.G560D variant in a cbEGF-like domain ( Table 2 ).
We compared the mutation type and age at event for patients with prophylactic aortic surgery versus dissection ( Table 1 ). Patients with dissection (n = 21) were less likely to have a missense mutation (14%) compared with patients who had prophylactic aortic surgery (n = 13; 31%; not statistically significant). The median age at the time of aortic event was younger for patients with truncating or splicing variants as compared with patients with missense variants. The median age for aortic dissection was 52 years in those with a missense mutation (n = 3), 40 years in those with a truncating mutation (n = 12), and 22 years in those with a splicing mutation (n = 5). A similar trend was observed in the prophylactic aortic surgery group ( Table 1 ). Overall, the median age for an aortic event was 51 years for patients with a missense mutation as compared with 29 years for patients with a truncating/splicing mutation.
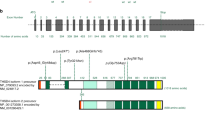
There were five different splicing variants in six of the probands in our aortic event cohort. In general, the mechanism whereby FBN1 splicing mutations exert their effect is unclear. Accordingly, we examined these five splicing variants to ascertain their potential impact on fibrillin-1 protein ( Table 3 ). Three of the variants (in four probands) were likely to create a frameshift and two of the novel splicing variants were predicted to cause exon skipping with an unknown impact on fibrillin-1. We identified two different splicing variants that did not occur in the universal GT splice donor or AG splice acceptor sites in three probands in the aortic event cohort. We observed an intron 11 variant, c.1468+5G>A, in two probands. Ogawa et al.3 found that this variant was associated with the activation of a latent splice donor site within exon 11, thereby creating a frameshift. The second intronic variant in the aortic event cohort that occurred outside of the GT or AG sites was c.4583-5A>G in intron 36. This variant was found to be a likely de novo variant in the proband (who fulfilled Ghent criteria) and therefore was likely pathogenic. Furthermore, five in silico splice prediction programs indicated that this alteration activates a cryptic splice acceptor site in intron 36 ( Table 3 ).
Four novel missense variants were identified in the aortic event cohort: p.C134Y, p.G560D, p.C587G, and p.Y2433C. The first three of these novel missense variants occurred at critical residues in the cbEGF-like domain for which different amino acid substitutions had been previously reported (p.C134G and p.C134S4,5; p.G560S6; and p.C587Y and p.C587R7,8). The fourth novel missense variant, p.Y2433C, also occurred in a cbEGF-like domain; it created an additional (seventh) cysteine residue, which likely disrupts fibrillin folding.
We observed three mutations (p.R1539X, p.R1541X, and c.1468+5G>A) that each occurred in two apparently unrelated patients ( Table 2 ). All three of these mutations have been observed numerous (9–12) times in the FBN1 Universal Mutation Database (http://www.umd.be/FBN1/; accessed 11 April 2014).
Discussion
The FBN1 gene is large, comprising 65 exons in which numerous alterations have been previously reported, but strong genotype–phenotype correlations have been elusive. Our data are novel in that we observed a high frequency of truncating and splicing FBN1 mutations in Ghent-positive patients with aortic events (aortic dissection and/or surgery) as compared with FBN1 mutation-positive patients without reported aortic events (79 vs. 39%, respectively; P < 0.0001). Considering only Ghent-positive patients, the difference was also significant: 48% of patients without reported aortic events had a truncating or splicing variant (versus 79% of Ghent-positive patients with an aortic event; P = 0.0039). We also observed that in patients who had an early aortic event, all or nearly all (12/12 (100%) of patients younger than age 30 and 20/21 (95%) of patients age 40 or younger) had a protein-truncating or -splicing variant. We characterized these age groups as having early aortic events because of a previous publication detailing aortic events in only 16% of men and 11% of women younger than 30 years, and ~40–45% of men and 25% of women younger than 40 years, in a large MFS cohort.1 We also observed that patients with aortic dissection had a higher frequency of a protein-truncating or -splicing variant (86%) as compared with patients with prophylactic aortic surgery (69%), although this observation was not statistically significant. In addition, we noted a trend in association between genotype and age at aortic surgery, dissection, or both. Overall, aortic dissection or surgery occurred at a younger age in patients with a truncating or splicing variant (median age, 29 years) as compared with patients with missense mutations (median age, 51 years).
Some previous publications have observed a nonsignificant trend toward aortic dissection occurring more frequently in patients with an FBN1 truncating or splicing mutation versus a missense or cysteine-substituting mutation.9,10 Specifically, Schrijver et al.10 found that dissection was the predominant indication for ascending aortic replacement in patients with a truncating mutation (58%; 15/26), whereas this was the case for only 32% (6/19) of patients with a missense mutation (P = 0.08). Rommel et al.9 likewise observed a nonsignificant trend: dissection occurred more often in their protein truncation group (5/25; 20%) than in their cysteine substitution group (3/30; 10%). A recent study observed an association with truncating and splicing mutations in Chinese patients with cardiovascular defects (dilatation, aneurysm, and dissection).11 Others have not reported an association with truncating mutations or cysteine mutations and cardiovascular manifestations.6,12,13,14 One reason that others may not have observed an association is because of the way the data were analyzed, for example, lumping dilatation and dissection and/or mitral valve prolapse together, including in the analysis patients who are too young to manifest cardiovascular events, and/or including patients who were older and more likely to manifest cardiovascular events. Détaint et al.1 observed a higher probability of ascending aortic dilatation, aortic event, and mitral valve prolapse in patients with mutations altering a cysteine residue. However, their published analysis did not split out aortic dissection or aortic surgery alone and genotype.
It is commonly thought that patients with a truncating mutation have a milder course of disease because of nonsense-mediated decay of the mutant transcript.15,16,17 Our data suggest that this is not the case, specifically our observation that the vast majority of early aortic events occurred in patients with a truncating (or splicing) variant. Therefore, instead of having a milder course of disease, as is commonly thought, patients with truncating or splicing variants may have a less clinically apparent MFS phenotype as compared with patients with missense variants, which also has been previously suggested by Schrijver et al.10 For example, one patient in our cohort was a 24-year-old woman with a splicing variant (predicted to result in protein truncation; c.4337-2A>G; Table 3 ) who died from an aortic dissection shortly after giving birth. When she was evaluated at the age of 12, she had some features of MFS but was considered not to have a diagnosis of MFS based on the Ghent criteria. The lack of diagnosis at age 12 may have been because of her young age, or it may have been because of her overall lack of clinically apparent MFS presentation, regardless of age.
The mechanism whereby FBN1 truncating or splicing mutations exert their effect is not totally clear, and both dominant negative and loss-of-function mechanisms have been identified. Although mechanistic studies of FBN1 splicing mutations have been performed on only a very limited basis, some FBN1 splicing mutations have been demonstrated to lead to a truncated protein, thereby having a similar impact as a nonsense or frameshift mutation.11,16,18,19 Other FBN1 splicing mutations cause in-frame exon deletions that disrupt microfibril assembly,20 theoretically having the same impact as a truncating mutation in a dominant negative effect model. Three of the five splicing variants in our cohort were likely to create a frameshift (leading to truncation), and two of the novel splicing variants were predicted to cause in-frame exon deletions with an unknown effect on fibrillin-1.
It has been observed that premature termination codon mutations in FBN1 can lead to preferentially degraded messenger RNA via nonsense-mediated messenger RNA decay pathways.15,16,21,22 Whereas rapid decay of the mutant transcript would be expected to create a loss-of-function phenotype, synthesis of a truncated transcript would be expected to create a dominant negative effect. This dominant negative effect would be due to integration of the truncated fibrillin-1 molecules into the microfibrils during microfibrillar assembly, thereby creating structurally inferior connective tissues. The dominant negative effect model was supported by the observation that most patients with classic MFS harbor missense mutations and the findings of patients with nonsense mutations and mild clinical presentation, in which they failed to meet the Ghent criteria.10,16,17 However, several others have stressed the importance of haploinsufficiency in the pathogenesis of MFS, as observed in mouse model studies and because of the identification of patients with classic MFS and full-gene deletions.23,24 It was also observed that variable disease expression in patients with premature termination codon mutations appeared to correlate with variable expression of the normal FBN1 allele and not with variable rates of nonsense-mediated decay.24
In recent years, it has come to light that the fibrillin-1-related mechanisms leading to MFS pathogenesis include not only its effect on extracellular matrix structure through microfibrillar formation and elastin association but also its effect on transforming growth factor (TGF)-β regulation and homeostasis. The discovery of Loeys-Dietz syndrome, a disorder phenotypically overlapping MFS (but generally with more aggressive aortic dilatation (and greater involvement of the arterial tree) and without ocular involvement), and mutations in genes encoding for TGF-β receptors (TGFBR1 and TGFBR2) helped to springboard the understanding of the importance of TGF-β signaling in MFS and related connective tissue disorders.25 It is now known that mutations in genes affecting MFS, Loeys-Dietz syndrome, and some other related connective tissue disorders lead to an increase in TGF-β signaling, and activation of both canonical (TGF-β/SMAD) and noncanonical (TGF-β/extracellular signal-regulated kinase (ERK)) pathways that have been shown to play a role in aneurysm development.26 These discoveries have also helped enhance the understanding of the efficacy of losartan (an angiotensin II type I receptor blocker) and RDEA-119 (an ERK antagonist) in attenuating aortic enlargement in MFS through the inhibition of TGF-β signaling.26,27,28 Whether FBN1 premature termination codon mutations have a stronger influence on TGF-β activation as compared with missense variants is unknown. The interplay between FBN1 mutation type and TGF-β signaling, dominant negative versus haploinsufficiency mechanisms, and dosage of normal fibrillin-1, as well as the effect on aneurysm progression and severity, deserves further investigation. Further elucidating these mechanisms has important potential for the treatment and management of patients with MFS and related disorders.
A limitation to our observations is related to the sometimes incomplete phenotypic information provided to us. Therefore, we cannot rule out the possibility that a patient with a pathogenic variant (missense, truncating, or splicing) who we categorized as not having a reported aortic event truly did not have aortic dissection/surgery. However, it should be noted that the frequency of aortic events in our cohort was similar to that reported by another larger study. Specifically, Détaint et al.1 observed a frequency of aortic events in 279 of 965 (28.9%) FBN1 mutation-positive patients ages 11 and older. We observed a similar aortic event frequency of 27.4% (34 of 124) in our cohort, considering patients ages 11 and older. Moreover, we were likely not biased by having a skewed percentage of mutation type because the missense mutation frequency (53.3%) in our overall cohort was similar to that observed in previous studies (50–59%).6,13,17 Furthermore, 18% of the patients in the cohort displayed mutations in exons 1–10, which is in agreement with previous works.12,16 Another potential limitation to our study is that our data may have been biased when younger or older patients were included in the analyses because of lower and higher frequencies of cardiovascular events, respectively. Furthermore, our data represent a mixture of ethnicities and therefore may unknowingly create a bias because of the potential for the presence of ethnicity-based differences in frequency of mutation type. Overall, it would be beneficial to use a more systematic method to better flesh out the association between protein-truncating/splicing variants and well-characterized cardiovascular features, including Z score, as well as other parameters such as patient age, sex, ethnicity, and presence of specific Ghent criteria features.
In summary, our findings are significant and novel in that they demonstrate a strong association of MFS patients with truncating/splicing FBN1 mutations with both age-independent and age-dependent (i.e., early) aortic events. We observed a novel trend among those patients who had an aortic event of the event occurring at a earlier median age in the presence of a truncating/splicing variant. Overall, the results of this study may have important implications for the management of patients with FBN1 truncating and splicing variants, especially in light of the fact that current practice generally considers such patients to have a more mild disease course. Our data suggest that although all patients with MFS may benefit from FBN1 gene sequencing for the purpose of confirming the diagnosis, affected individuals should be monitored similarly for aortic dilatation and its complications, regardless of the FBN1 mutation type.
Disclosure
The authors declare no conflict of interest.
References
Détaint D, Faivre L, Collod-Beroud G, et al. Cardiovascular manifestations in men and women carrying a FBN1 mutation. Eur Heart J 2010;31:2223–2229.
Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet 2010;47:476–485.
Ogawa N, Imai Y, Takahashi Y, et al. Evaluating Japanese patients with the Marfan syndrome using high-throughput microarray-based mutational analysis of fibrillin-1 gene. Am J Cardiol 2011;108:1801–1807.
Howarth R, Yearwood C, Harvey JF . Application of dHPLC for mutation detection of the fibrillin-1 gene for the diagnosis of Marfan syndrome in a National Health Service Laboratory. Genet Test 2007;11:146–152.
Sakai H, Visser R, Ikegawa S, et al. Comprehensive genetic analysis of relevant four genes in 49 patients with Marfan syndrome or Marfan-related phenotypes. Am J Med Genet A 2006;140:1719–1725.
Loeys B, Nuytinck L, Delvaux I, De Bie S, De Paepe A . Genotype and phenotype analysis of 171 patients referred for molecular study of the fibrillin-1 gene FBN1 because of suspected Marfan syndrome. Arch Intern Med 2001;161:2447–2454.
Booms P, Cisler J, Mathews KR, et al. Novel exon skipping mutation in the fibrillin-1 gene: two ‘hot spots’ for the neonatal Marfan syndrome. Clin Genet 1999;55:110–117.
Liang C, Fan W, Wu S, Liu Y . Identification of a novel FBN1 mutation in a Chinese family with isolated ectopia lentis. Mol Vis 2011;17:3481–3485.
Rommel K, Karck M, Haverich A, et al. Identification of 29 novel and nine recurrent fibrillin-1 (FBN1) mutations and genotype-phenotype correlations in 76 patients with Marfan syndrome. Hum Mutat 2005;26:529–539.
Schrijver I, Liu W, Odom R, et al. Premature termination mutations in FBN1: distinct effects on differential allelic expression and on protein and clinical phenotypes. Am J Hum Genet 2002;71:223–237.
Wang WJ, Han P, Zheng J, et al. Exon 47 skipping of fibrillin-1 leads preferentially to cardiovascular defects in patients with thoracic aortic aneurysms and dissections. J Mol Med (Berl) 2013;91:37–47.
Comeglio P, Johnson P, Arno G, et al. The importance of mutation detection in Marfan syndrome and Marfan-related disorders: report of 193 FBN1 mutations. Hum Mutat 2007;28:928.
Arbustini E, Grasso M, Ansaldi S, et al. Identification of sixty-two novel and twelve known FBN1 mutations in eighty-one unrelated probands with Marfan syndrome and other fibrillinopathies. Hum Mutat 2005;26:494.
Loeys B, De Backer J, Van Acker P, et al. Comprehensive molecular screening of the FBN1 gene favors locus homogeneity of classical Marfan syndrome. Hum Mutat 2004;24:140–146.
Aoyama T, Francke U, Dietz HC, Furthmayr H . Quantitative differences in biosynthesis and extracellular deposition of fibrillin in cultured fibroblasts distinguish five groups of Marfan syndrome patients and suggest distinct pathogenetic mechanisms. J Clin Invest 1994;94:130–137.
Dietz HC, McIntosh I, Sakai LY, et al. Four novel FBN1 mutations: significance for mutant transcript level and EGF-like domain calcium binding in the pathogenesis of Marfan syndrome. Genomics 1993;17:468–475.
Rand-Hendriksen S, Tjeldhorn L, Lundby R, et al. Search for correlations between FBN1 genotype and complete Ghent phenotype in 44 unrelated Norwegian patients with Marfan syndrome. Am J Med Genet A 2007;143A:1968–1977.
Guo D, Tan FK, Cantu A, Plon SE, Milewicz DM . FBN1 exon 2 splicing error in a patient with Marfan syndrome. Am J Med Genet 2001;101:130–134.
Dietz HC, Valle D, Francomano CA, Kendzior RJ Jr, Pyeritz RE, Cutting GR . The skipping of constitutive exons in vivo induced by nonsense mutations. Science 1993;259:680–683.
Liu W, Qian C, Comeau K, Brenn T, Furthmayr H, Francke U . Mutant fibrillin-1 monomers lacking EGF-like domains disrupt microfibril assembly and cause severe marfan syndrome. Hum Mol Genet 1996;5:1581–1587.
Schrijver I, Liu W, Brenn T, Furthmayr H, Francke U . Cysteine substitutions in epidermal growth factor-like domains of fibrillin-1: distinct effects on biochemical and clinical phenotypes. Am J Hum Genet 1999;65:1007–1020.
Karttunen L, Ukkonen T, Kainulainen K, Syvänen AC, Peltonen L . Two novel fibrillin-1 mutations resulting in premature termination codons but in different mutant transcript levels and clinical phenotypes. Hum Mutat 1998(suppl 1):S34–S37.
Hilhorst-Hofstee Y, Hamel BC, Verheij JB, et al. The clinical spectrum of complete FBN1 allele deletions. Eur J Hum Genet 2011;19:247–252.
Hutchinson S, Furger A, Halliday D, et al. Allelic variation in normal human FBN1 expression in a family with Marfan syndrome: a potential modifier of phenotype? Hum Mol Genet 2003;12:2269–2276.
Loeys BL, Chen J, Neptune ER, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet 2005;37:275–281.
Holm TM, Habashi JP, Doyle JJ, et al. Noncanonical TGFβ signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science 2011;332:358–361.
Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz HC 3rd . Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. N Engl J Med 2008;358:2787–2795.
Nataatmadja M, West J, Prabowo S, West M . Angiotensin II receptor antagonism reduces transforming growth factor beta and Smad signaling in thoracic aortic aneurysm. Ochsner J 2013;13:42–48.
Comeglio P, Evans AL, Brice G, Cooling RJ, Child AH . Identification of FBN1 gene mutations in patients with ectopia lentis and marfanoid habitus. Br J Ophthalmol 2002;86:1359–1362.
Hayward C, Porteous ME, Brock DJ . Mutation screening of all 65 exons of the fibrillin-1 gene in 60 patients with Marfan syndrome: report of 12 novel mutations. Hum Mutat 1997;10:280–289.
Nijbroek G, Sood S, McIntosh I, et al. Fifteen novel FBN1 mutations causing Marfan syndrome detected by heteroduplex analysis of genomic amplicons. Am J Hum Genet 1995;57:8–21.
Hung CC, Lin SY, Lee CN, et al. Mutation spectrum of the fibrillin-1 (FBN1) gene in Taiwanese patients with Marfan syndrome. Ann Hum Genet 2009;73(Pt 6):559–567.
Hewett DR, Lynch JR, Smith R, Sykes BC . A novel fibrillin mutation in the Marfan syndrome which could disrupt calcium binding of the epidermal growth factor-like module. Hum Mol Genet 1993;2:475–477.
Mátyás G, De Paepe A, Halliday D, Boileau C, Pals G, Steinmann B . Evaluation and application of denaturing HPLC for mutation detection in Marfan syndrome: Identification of 20 novel mutations and two novel polymorphisms in the FBN1 gene. Hum Mutat 2002;19:443–456.
Youil R, Toner TJ, Bull E, et al. Enzymatic mutation detection (EMD) of novel mutations (R565X and R1523X) in the FBN1 gene of patients with Marfan syndrome using T4 endonuclease VII. Hum Mutat 2000;16:92–93.
Tiecke F, Katzke S, Booms P, et al. Classic, atypically severe and neonatal Marfan syndrome: twelve mutations and genotype-phenotype correlations in FBN1 exons 24-40. Eur J Hum Genet 2001;9:13–21.
Halliday D, Hutchinson S, Kettle S, Firth H, Wordsworth P, Handford PA . Molecular analysis of eight mutations in FBN1. Hum Genet 1999;105:587–597.
Halliday DJ, Hutchinson S, Lonie L, et al. Twelve novel FBN1 mutations in Marfan syndrome and Marfan related phenotypes test the feasibility of FBN1 mutation testing in clinical practice. J Med Genet 2002;39:589–593.
Körkkö J, Kaitila I, Lönnqvist L, Peltonen L, Ala-Kokko L . Sensitivity of conformation sensitive gel electrophoresis in detecting mutations in Marfan syndrome and related conditions. J Med Genet 2002;39:34–41.
Stheneur C, Collod-Béroud G, Faivre L, et al. Identification of the minimal combination of clinical features in probands for efficient mutation detection in the FBN1 gene. Eur J Hum Genet 2009;17:1121–1128.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Baudhuin, L., Kotzer, K. & Lagerstedt, S. Increased frequency of FBN1 truncating and splicing variants in Marfan syndrome patients with aortic events. Genet Med 17, 177–187 (2015). https://doi.org/10.1038/gim.2014.91
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2014.91
