
Abstract
Purpose:
The aim of this study was to compare guidance for genetic testing in US Food and Drug Administration (FDA)-approved drug labels in oncology to those of drugs for other therapeutic areas.
Methods:
We reviewed labels of all the FDA-approved drugs with labels containing pharmacogenomic information. We assessed whether genetic testing was required or recommended before prescription and, if not, the reason for pharmacogenomic labeling.
Results:
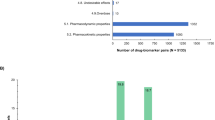
We included 140 drugs corresponding to 158 drug–biomarker pairs. Overall, 46 (29%) of 158 pairs stated a requirement or recommendation for genetic biomarker testing in the label. This proportion was higher in oncology than in other areas (62 vs. 12%; P < 0.001). For the 112 drug–biomarker pairs (including 20 in oncology) without recommendation or requirement for genetic testing, the main reasons for pharmacogenomic labeling were change in pharmacologic end points (32%) and higher risk of toxicity (30%). For 11 (10%) pairs (including 1 in oncology), a genetic biomarker was mentioned only to inform that it was not relevant. In oncology, the main reasons for pharmacogenomic labeling were higher risk of toxicity (55%) and definition of the mechanism of action (25%).
Conclusion:
Inclusion of biomarkers in drug labels does not always correspond to required or recommended genetic testing, especially outside oncology.
Genet Med 17 9, 733–738.
Similar content being viewed by others


Introduction
Pharmacogenomics studies the role of inherited and acquired genetic variation in drug response in order to tailor treatment to individual genetic and genomic characteristics.1 This field is a sizable, and perhaps the most actionable, part of personalized medicine.2 Several pharmacogenomic biomarkers are used to predict drug efficacy or higher risk of safety issues such as those involving human leukocyte antigen B and hypersensitivity to abacavir,3 UGT1A1 and irinotecan,4 tumor gene expression and breast cancer,5 or cytochrome P450 (CYP450) polymorphisms and patients treated by selective serotonin reuptake inhibitors.6
Since 2009, the US Food and Drug Administration (FDA) has listed the drugs whose labels mention pharmacogenomic information.7 This number is increasing, and this has been used as a marker of success and development of personalized medicine.8 However, the mention of a pharmacogenomic biomarker in the drug label can have different meanings depending on the drug, and thus very different implications for clinicians and patients. For instance, the information can mean that genetic testing is required or recommended to define the population to treat. However, it can also mean that a high risk of side effects has been reported for some biomarker-based subpopulations, or simply that the pharmacogenomic biomarker is involved in the drug mechanism of action. Genetic testing is not required in the latter two cases, but prescribers of the drug lack a clear course of action. In such cases, this genetic information may not be relevant to personalized medicine.
Previous studies have reported a low frequency of requirement or recommendation for genetic testing among drugs with a pharmacogenomic biomarker mentioned in the label.9,10 However, they did not stratify analyses by therapeutic area. New oncology drugs are often targeted therapies, so we could expect that genetic testing for oncology drugs would be required or at least recommended to define the population to treat. Here, we aimed to compare guidance for genetic (pharmacogenomic biomarker) testing in drug labels for FDA-approved oncology and nononcology drugs.
Materials and Methods
List of drugs
The Table of Pharmacogenomic Biomarkers in Drug Labeling on the FDA website7 (hereafter the FDA table) includes all drugs with corresponding biomarkers listed in the label. One or several biomarkers can be listed for a given drug, leading in the latter case to different drug–biomarker pairs. Each drug in the FDA table is linked to one or several entries in the Drugs@FDA database, which lists all available drug applications: new drug applications, biologic license applications (for biologic agents), and abbreviated new drug applications (for generic drugs), as well as corresponding labels.
Data extraction
The following information was extracted from the FDA table on 20 June 2014 and imported into a database: name of the drug; therapeutic area (oncology or nononcology, drugs classified in both oncology and another therapeutic area were classified in oncology); gene involved in the biomarker; referenced subgroup defined by the biomarker; and label sections containing pharmacogenomic biomarker information.
One investigator (A.V.) used a standardized form to extract the following information from the Drugs@FDA database for each drug–biomarker pair: date of first approval of the drug, date of first mention of the genetic information in the label, mention of genetic information at approval, mention of genetic information in a black box warning (the strongest warning that can be issued by the FDA),11 existence of at least one biomarker-based indication, and existence of at least one biomarker-based contraindication.
A drug can have several approval dates: one for each application submitted for FDA review. We were interested in the first approval date for the drug (as a new molecular entity, new active ingredient, new combination, or drug already marked without an approved new drug application). If a given drug from the FDA table was linked to an application for a drug whose chemical type was not a new agent (i.e., new dosage form, new formulation or new manufacturer, or new indication), we searched the Drugs@FDA database with the corresponding “Active Ingredient(s)” as search term. Among all retrieved entries, we selected the entry with “1 New Molecular Entity” as the chemical type and used the corresponding approval date. If no entries were characterized as new molecular entities, we used the oldest date of approval. Because this classification does not exist for biologic license applications, we performed the same search for all drugs approved with a biologic license application to ensure that we extracted the oldest approval date.
We considered pharmacogenomic labeling to be present at approval if we could assess the drug label at the time of approval and if pharmacogenomic information was present in this label. If we could not assess the label (especially for older drugs), we considered it to be missing data for this variable. Nonetheless, we considered that pharmacogenomic labeling was not possible at approval for CYP450s and drugs approved before the seminal article of Cooper et al., which was published in 1965.12 Similarly, for glucose-6-phosphate dehydrogenase, we considered that pharmacogenomic labeling was not present at approval for drugs approved before the publication by Alving and colleagues in 1956.13
Characteristics of pharmacogenomic labeling
We assigned to each drug–biomarker pair a category of guidance for genetic testing built on the Pharmacogenomics Knowledgebase (PharmGKB) classification.14 The PharmGKB is an academic-based (Stanford University) collaboration whose curators read labels of drugs to assign them a “PGx [Pharmacogenomics] level.” One author (A.V.), blinded to PharmGKB results, read drug labels and assessed whether genetic testing was required (PharmGKB corresponding category: “Genetic testing required”), recommended (“Genetic testing recommended”), or neither required nor recommended (“Actionable PGx” and “Informative PGx”). Examples of classification of selected drug–biomarker pairs to categories of guidance are provided in Table 1 . The agreement between our results and the corresponding evaluation displayed on the PharmGKB Web site (after having regrouped the “Actionable PGx” and “Informative PGx” categories) was good (Cohen’s κ = 0.79; 95% confidence interval: 0.67–0.88). All discordances were reviewed by a second investigator (R.P.), and consensus was reached for all. After completing the consensus process, eight pairs remain discordant with PharmGKB classification. A list of those eight pairs with comments about discordances is provided in Supplementary Table S1 online.
For drug–biomarker pairs without a genetic testing requirement or recommendation in the label, two investigators (A.V. and R.P.) independently extracted the justification for pharmacogenomic labeling and categorized it using the following classifications we developed:
-
1. The biomarker is used to define the drug mechanism of action.
-
2. The biomarker is used to define one medical condition for which the drug is indicated.
-
3. Drug interactions with drugs metabolized by a biomarker-related enzyme (e.g., CYP450) have been reported or are expected.
-
4. Variations in pharmacologic end points based on biomarker-based subpopulations have been reported.
-
5. Increased toxicity (on nonpharmacologic end points) has been reported in one biomarker-based subpopulation.
-
6. Decreased efficacy (on nonpharmacologic end points) has been reported in one biomarker-based subpopulation.
-
7. The biomarker is mentioned in the label only to inform that it is not relevant to patients and caregivers.
Discordances between the two investigators were resolved by consensus. Examples of drug–biomarker pairs for each level of classification are displayed in Table 2 .
Drug approval by year and therapeutic area
To determine whether pharmacogenomic labeling was more frequent in oncology than in other therapeutic areas, we compared the proportion of drugs approved for use in oncology among drugs with pharmacogenomic labeling for 1996 to 2013 with the proportion of drugs approved for use in oncology among all new molecular entities approved by the FDA during the same period. To do so, we used the Drugs@FDA database, which reports all regulatory actions by the FDA, downloaded on 19 March 2014. The therapeutic area was identified by using Anatomical Therapeutic Chemical codes15 (for exact definitions and methods used for Anatomical Therapeutic Chemical mapping, see the Supplementary Materials and Methods online).
Statistical analysis
We stratified all analyses by therapeutic area (oncology versus nononcology). Continuous variables are presented as medians and interquartile ranges, and qualitative variables are presented as frequencies and percentages. Proportions were compared between oncology and nononcology drugs by Fisher’s exact tests. Distributions of continuous variables were compared by Mann–Whitney–Wilcoxon tests. All tests were performed with a type I error rate of 5% and the R statistical software version 3.1.16
Results
Characteristics of drugs and drug–biomarker pairs
We removed three duplicates of the 161 drug–biomarker pairs on the FDA website. Hence, we included 158 drug–biomarker pairs corresponding to 140 different drugs. In total, 126 drugs had only one biomarker; 11 drugs had two biomarkers; 2 drugs had three biomarkers (ESR1-PGR, F5, and F2, for tamoxifen; and VKORC1, CYP2C9, and PROC, for warfarin); and 1 drug (imatinib) had four biomarkers (KIT, BCR/ABL1, PDGFRB, and FIP1L1/PDGFRA). Overall, 30 drugs had at least one biomarker-based indication and six had a biomarker-based contraindications ( Table 3 ). One-third of the drug–biomarker pairs (n = 53, 34%) concerned drugs indicated for oncology, corresponding to 42 unique drugs.
Comparison between oncology and nononcology drugs
For the 53 oncology drug–biomarker pairs, the five most frequent corresponding genes were BCR/ABL1 (n = 6), EGFR (n = 5), HER2 (n = 5), MS4A1 (n = 4), and BRAF (n = 3). For the 105 nononcology drug–biomarker pairs, the five most frequent corresponding genes were CYP2D6 (n = 37), G6PD (n = 17), CYP2C19 (n = 15), IFNL3 (n = 5), and LDLR (n = 5). CYP450 enzymes represented the majority (n = 56, 53%) of nononcology drug–biomarker pairs.
Requirement or recommendation for genetic testing was significantly more frequent for oncology drug–biomarker pairs as compared with nononcology drug–biomarker pairs. Of the 53 oncology drug–biomarker pairs, genetic biomarker testing was required for 33 (59%) and recommended for 0 (0%), as compared with 9 (9%) and 4 (4%), respectively, of the 105 nononcology drug–biomarker pairs (P < 0.001). Supplementary Tables S2 and S3 online display the main characteristics of drug–biomarker pairs with a requirement or recommendation for genetic testing for oncology and nononcology drugs, respectively. Pharmacogenomics was used to define at least one indication for 27 of 42 (64%) oncology drugs as compared with only 3 of 98 (3%) nononcology drugs (P < 0.001). Nonetheless, the proportion of drugs with at least one biomarker-based contraindication was similar for nononcology and oncology drugs (5 vs. 4%, respectively; P = 1). Pharmacogenetic information was present at approval for 31 of 53 oncology drug–biomarker pairs (66%, 6 missing data) and for 49 of 105 nononcology pairs (61%, 25 missing data; P = 0.70).
For drugs without requirement or recommendation for genetic testing, pharmacogenomic labeling often described changes in pharmacologic end points (n = 36; 32%), such as increased exposure to the drug as measured by the area under the curve or increased clearance of the drug; or increased toxicity (n = 34, 30%), such as high expected rates of adverse events or contraindication in patients with known genetic deficiency but still without a clear statement regarding genetic testing ( Table 3 ). Of note, for 10% of drug–biomarker pairs, the genetic biomarker was mentioned only to inform that it was not relevant for the drug use. For oncology drugs, half of the labels (n = 11, 55%) concerned increased toxicity; for five drugs (25%), a genetic biomarker was mentioned to define the drug’s mechanism of action (e.g., an anti-CD30 drug without biomarker-based indication).
Proportion of drugs approved for use in oncology among drugs with pharmacogenomic labeling from 1996 to 2013
From the Drugs@FDA database, we identified 514 new drugs that were approved from 1996 to 2013. In total, 75 (15%) were indicated for oncology, and the proportion of drug–biomarker pairs with pharmacogenomic labeling that had oncology indications during the same time was 53/150 (35%). Moreover, during the 2003–2013 period, 20 of 54 (37%) new oncology drugs had pharmacogenomic labeling at approval as compared with 20 of 225 (9%) new nononcology drugs.
Discussion
In this study, we showed that oncology drugs represent one-third of the drugs with pharmacogenomic labeling. Furthermore, we found that a high proportion of pharmacogenomic labeling for oncology drugs required or recommended genetic biomarker testing, contrary to nononcology drugs. This result may contrast with the global picture (that most pharmacogenomic labeling does not require or recommend genetic testing) reported in previous studies.9,10 A likely explanation for the predominance of oncology and for the high proportion of genetic testing requirements or recommendations in oncology is the high number of targeted drugs in this area. These drugs are often tested in enrichment trials that are restricted to patients who are positive for the biomarker, which is often a somatic mutation (e.g., BRAF V600E/K mutations in melanoma). Hence, the indications section of the drug label mentions the biomarker testing to be consistent with the inclusion criteria of trials that have supported the drug approval. Pharmacogenomic labeling could be considered frequent in oncology. However, in the past decade, less than half of new approved oncology drugs had pharmacogenomic labeling at approval.
Our results are concordant with recent work focusing on content of patient-targeted sections for the 140 drugs included in the FDA table.17 This work showed that only 29 drug labels (21%) mentioned pharmacogenetic information for patients, and the authors speculated that the reason for this low frequency may be that “the information is considered unnecessary for patients to use the drug safely and effectively.”17 Indeed, as underlined in our study, for the majority of drugs the pharmacogenetic information in drug labels does not translate into a specific course of action for patients and prescribers.
When genetic testing was not required or recommended in the drug label, the genetic biomarker listed was often related to the drug metabolism (e.g., CYP450, UGT1A1, DPYD, TPMT), and the label described changes in pharmacologic end points or increased toxicity. Requirements or recommendations for genetic testing in these cases were rare because genotypes or efficacy associations were less strong than for targeted drugs. Many nongenetic causes of phenotypic variability (e.g., age, sex, concomitant drug use, body weight, renal function, comorbidities, and phenoconversion)18,19 can explain why genotypes sometimes do not accurately predict drug response. Warfarin and clopidogrel are two examples for which pharmacogenomic biomarkers may not be clinically useful, contrary to previous expectations.20,21
One limitation of our study is that we studied only FDA-approved drugs. Although we lack an official list of drugs with biomarker information in the label for countries other than the United States, some authors found discrepancies between the United States, Europe, and Japan in the number of such drugs and the contents of the labels.22,23,24 Moreover, the phrasing and the number of indications for a given drug may differ between the United States and Europe.25 Shimazawa et al.23 found substantial discordances in labels between Japan, the United States, and the United Kingdom, but these discordances were low for the indications section of labels and for biomarkers with recommendations for genetic testing. Therefore, our results should not be directly extrapolated outside the United States; however, these differences are not likely to change our main conclusion.
We found that genetic testing is required or recommended by health authorities before prescription for the majority of oncology drugs with pharmacogenomic labeling, but for less than one-fifth of nononcology drugs. Nonetheless, our results do not indicate that a high level of evidence exists to show that genetic testing improves clinically relevant outcomes in these situations (that is, demonstration of clinical utility).26 For instance, a drug that has been tested only in a biomarker-based subpopulation would have a requirement or a recommendation for genetic testing, but this does not mean that the drug is not effective for other patients. A recent study evaluating the level of evidence for clinical validity (the ability to predict phenotype) and utility of drug–biomarker pairs in an earlier extraction of the FDA table showed that the labels contained convincing evidence of clinical validity for 43 of the 119 pairs (36%) and of clinical utility for 18 pairs only (15%).27 This underlines a lack of studies for proper evaluation of the clinical validity and utility of biomarkers despite pharmacogenomic information mentioned in drug labels.
Proper validation of the clinical utility of a biomarker can be assessed with a prospective design in a randomized controlled trial comparing a biomarker-based strategy with a treat-all strategy.28,29 However, this design would not be suitable for many situations. Another method is to retrospectively assess biomarkers in clinical data from observational studies or randomized controlled trials in which treatment received did not depend on the biomarker to compare the risk–benefit ratio according to biomarker values.30,31 Several organizations, such as the Evaluation of Genomic Applications in Practice and Prevention Working Group sponsored by the Centers for Disease Control and Prevention,32 the Clinical Pharmacogenetics Implementation Consortium, and other groups, are currently conducting research to address this crucial question of the clinical utility of genetic biomarkers.
Concerning the usefulness of the global approach of selecting oncology drugs based on genetic abnormalities rather than pathological features, at least two clinical trials, the SHIVA trial in France33,34 and the NCI-MPACT in the United States35 are comparing a biomarker-based therapy (molecular tumor profiling and corresponding targeted therapies) with conventional therapy for patients with advanced solid cancer of any type.
Conclusion
From information contained in drug labels, we found a high proportion of oncology drug–biomarker pairs for which testing was required or recommended. Conversely, for most nononcology drug–biomarker pairs, genetic testing is neither required nor recommended before drug prescription, and therefore most of these pharmacogenetic labels do not translate into a specific course of action for patients and prescribers. Impacts of genetic biomarker development are clearly visible in oncology drug labels, but hopes of personalizing treatment based on genetic biomarkers in other therapeutic areas have not yet been fulfilled.
Disclosure
R.P. reports receiving financial compensation for consultancies from Roche and Amgen. The other authors declare no conflict of interest.
References
Wang L, McLeod HL, Weinshilboum RM . Genomics and drug response. N Engl J Med 2011;364:1144–1153.
Scott SA . Personalizing medicine with clinical pharmacogenetics. Genet Med 2011;13:987–995.
Mallal S, Phillips E, Carosi G, et al.; PREDICT-1 Study Team. HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med 2008;358:568–579.
Palomaki GE, Bradley LA, Douglas MP, Kolor K, Dotson WD . Can UGT1A1 genotyping reduce morbidity and mortality in patients with metastatic colorectal cancer treated with irinotecan? An evidence-based review. Genet Med 2009;11:21–34.
Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group. Recommendations from the EGAPP Working Group: can tumor gene expression profiling improve outcomes in patients with breast cancer? Genet Med 2009;11:66–73.
Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group. Recommendations from the EGAPP Working Group: testing for cytochrome P450 polymorphisms in adults with nonpsychotic depression treated with selective serotonin reuptake inhibitors. Genet Med 2007;9:819–825.
US Food and Drug Administration. Genomics - Table of Pharmacogenomic Biomarkers in Drug Labeling. http://www.fda.gov/drugs/scienceresearch/researchareas/pharmacogenetics/ucm083378.htm. Accessed 20 June 2014.
Abrahams E, Silver M . The case for personalized medicine. J Diabetes Sci Technol 2009;3:680–684.
Tutton R . Pharmacogenomic biomarkers in drug labels: what do they tell us? Pharmacogenomics 2014;15:297–304.
Ioannidis JP, Khoury MJ . Are randomized trials obsolete or more important than ever in the genomic era? Genome Med 2013;5:32.
US Food and Drug Administration. Guidance for industry: warnings and precautions, contraindications, and boxed warning sections of labeling for human prescription drug and biological products—content and format. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm075096.pdf. Accessed 20 June 2014.
Cooper DY, Levin S, Narasimhulu S, Rosenthal O . Photochemical action spectrum of the terminal oxidase of mixed function oxidase systems. Science 1965;147:400–402.
Carson PE, Flanagan CL, Ickes CE, Alving AS . Enzymatic deficiency in primaquine-sensitive erythrocytes. Science 1956;124:484–485.
PharmGKB. Drug Label Legend. http://www.pharmgkb.org/page/drugLabelLegend. Accessed 20 June 2014.
World Health Organization (WHO) Collaborating Center for Drug Statistics Methodology. ATC classification index with DDDs, 2014. 2014. http://www.whocc.no/atc_ddd_index/. Accessed 24 March, 2014.
R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria, 2013. http://www.R-project.org/. Accessed 21 March, 2014.
Haga S, Mills R, Moaddeb J . Pharmacogenetic information for patients on drug labels. Pharmgenomics Pers Med 2014;7:297–305.
Sadée W, Dai Z . Pharmacogenetics/genomics and personalized medicine. Hum Mol Genet 2005;14 Spec No. 2:R207–R214.
Preskorn SH, Kane CP, Lobello K, et al. Cytochrome P450 2D6 phenoconversion is common in patients being treated for depression: implications for personalized medicine. J Clin Psychiatry 2013;74:614–621.
Stergiopoulos K, Brown DL . Genotype-guided vs clinical dosing of warfarin and its analogues: meta-analysis of randomized clinical trials. JAMA Intern Med 2014;174:1330–1338.
Osnabrugge RL, Head SJ, Zijlstra F, et al. A systematic review and critical assessment of 11 discordant meta-analyses on reduced-function CYP2C19 genotype and risk of adverse clinical outcomes in clopidogrel users. Genet Med 2014; e-pub ahead of print 19 June 2014.
Malottki K, Biswas M, Deeks JJ, et al. Stratified medicine in European Medicines Agency licensing: a systematic review of predictive biomarkers. BMJ Open 2014;4:e004188.
Shimazawa R, Ikeda M . Differences in pharmacogenomic biomarker information in package inserts from the United States, the United Kingdom and Japan. J Clin Pharm Ther 2013;38:468–475.
Shimazawa R, Ikeda M . Approval gap of pharmacogenomic biomarkers and in vitro companion diagnostics between the United States and Japan. J Clin Pharm Ther 2014;39:210–214.
Trotta F, Leufkens HG, Schellens JH, Laing R, Tafuri G . Evaluation of oncology drugs at the European Medicines Agency and US Food and Drug Administration: when differences have an impact on clinical practice. J Clin Oncol 2011;29:2266–2272.
Lesko LJ, Zineh I, Huang SM . What is clinical utility and why should we care? Clin Pharmacol Ther 2010;88:729–733.
Wang B, Canestaro WJ, Choudhry NK . Clinical evidence supporting pharmacogenomic biomarker testing provided in US Food and Drug Administration drug labels. JAMA Intern Med 2014; e-pub ahead of print 13 October 2014.
Tajik P, Zwinderman AH, Mol BW, Bossuyt PM . Trial designs for personalizing cancer care: a systematic review and classification. Clin Cancer Res 2013;19:4578–4588.
Mandrekar SJ, Sargent DJ . Clinical trial designs for predictive biomarker validation: theoretical considerations and practical challenges. J Clin Oncol 2009;27:4027–4034.
Janes H, Pepe MS, Bossuyt PM, Barlow WE . Measuring the performance of markers for guiding treatment decisions. Ann Intern Med 2011;154:253–259.
Simon RM, Paik S, Hayes DF . Use of archived specimens in evaluation of prognostic and predictive biomarkers. J Natl Cancer Inst 2009;101:1446–1452.
Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group. The EGAPP initiative: lessons learned. Genet Med 2014;16:217–224.
Le Tourneau C, Paoletti X, Servant N, et al. Randomised proof-of-concept phase II trial comparing targeted therapy based on tumour molecular profiling vs conventional therapy in patients with refractory cancer: results of the feasibility part of the SHIVA trial. Br J Cancer 2014;111:17–24.
Le Tourneau C, Kamal M, Trédan O, et al. Designs and challenges for personalized medicine studies in oncology: focus on the SHIVA trial. Target Oncol 2012;7:253–265.
Kummar S, Williams M, Lih C-J, et al. NCI MPACT: National Cancer Institute molecular profiling-based assignment of cancer therapy [abstract]. J Clin Oncol 2014;32:5s.
Acknowledgements
This work was supported in part by an academic grant from the program “Équipe Espoir de la Recherche,” Fondation pour la Recherche Médicale, Paris, France (DEQ20101221475). We thank Laura Smales (BioMedEditing, Toronto, Canada) and Caroline G. Barnes for proofreading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Tables
(DOC 76 kb)
Supplementary Materials and Methods
(DOC 33 kb)
Rights and permissions
About this article

Cite this article
Vivot, A., Boutron, I., Ravaud, P. et al. Guidance for pharmacogenomic biomarker testing in labels of FDA-approved drugs. Genet Med 17, 733–738 (2015). https://doi.org/10.1038/gim.2014.181
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2014.181
Keywords
This article is cited by
-
Classification of Companion Diagnostics: A New Framework for Biomarker-Driven Patient Selection
Therapeutic Innovation & Regulatory Science (2022)
-
Pharmacogenomic biomarker information differences between drug labels in the United States and Hungary: implementation from medical practitioner view
The Pharmacogenomics Journal (2020)
-
Evidence for Treatment-by-Biomarker interaction for FDA-approved Oncology Drugs with Required Pharmacogenomic Biomarker Testing
Scientific Reports (2017)
-
Comparing cytochrome P450 pharmacogenetic information available on United States drug labels and European Union Summaries of Product Characteristics
The Pharmacogenomics Journal (2017)
