
Abstract
Purpose:
Genetic testing is routinely used for second-tier confirmation of newborn sequencing results to rule out false positives and to confirm diagnoses in newborns undergoing inpatient and outpatient care. We developed a targeted next-generation sequencing panel coupled with a variant processing pipeline and demonstrated utility and performance benchmarks across multiple newborn disease presentations in a retrospective clinical study.
Methods:
The test utilizes an in silico gene filter that focuses directly on 126 genes related to newborn screening diseases and is applied to the exome or a next-generation sequencing panel called NBDx. NBDx targets the 126 genes and additional newborn-specific disorders. It integrates DNA isolation from minimally invasive biological specimens, targeted next-generation screening, and rapid characterization of genetic variation.
Results:
We report a rapid parallel processing of 8 to 20 cases within 105 hours with high coverage on our NBDx panel. Analytical sensitivity of 99.8% was observed across known mutation hotspots. Concordance calls with or without clinical summaries were 94% and 75%, respectively.
Conclusion:
Rapid, automated targeted next-generation sequencing and analysis are practical in newborns for second-tier confirmation and neonatal intensive care unit diagnoses, laying a foundation for future primary DNA-based molecular screening of additional disorders and improving existing molecular testing options for newborns.
Genet Med 17 5, 337–347.
Similar content being viewed by others


Introduction
Of the approximately 4,000 single-gene disorders (Mendelian diseases) with a known molecular basis,1 a significant fraction manifest symptoms during the newborn period (first 28 days of life). Newborn screening (NBS) programs administer an infant’s first biochemical screening test from a dried blood spot (DBS) specimen for 30 to 50 severe genetic disorders for which public health interventions exist, and thus these programs are successful in preventing mortality or life-long debilitation.2 However, positive results require complex second-tier confirmation to address false-positive results. For example, in 2007, of the 3,364,612 NBS primary screenings, 1,249 were reported positive for maple syrup urine disease (OMIM 248600), but only 18 actually had the disease.3
For neonates with genetic disorders, a rapid diagnosis of newborn diseases could make the difference between life and death and reduce length of stay in the neonatal intensive care unit (NICU). However, in modern medical practice acutely ill newborns are stabilized in the NICU and discharged without a genetic diagnosis. The burden of genetic disorders is estimated at upwards of 25% of inpatient admissions in the newborn and pediatric population.4,5 Previously, genetic testing was performed gene by gene, based on available clinical indications and family histories, with each test conducted serially and costing thousands of dollars. With the advent of next-generation sequencing (NGS), large panels of genes can now be scanned together rapidly at a lower cost and with the added promise of reduced length of stay and better outcomes.
Genome-scale technologies such as whole-genome sequencing (WGS) and whole-exome sequencing (WES) have been proposed for newborn and pediatric diagnostic medical care.2,6,7,8,9,10,11 The US National Institutes of Health funded four studies for $25 million to determine the ethical implications of WGS and WES in newborns and their potential utility.7 Still, several bottlenecks limit immediate and wider adoption of NGS testing for newborns. First, the turnaround time for current commercial genetic testing may take weeks or months to generate a clinical report, which is impractical for guiding the clinical care of acutely ill newborns. Second, minimally invasive NGS testing methods appropriate for newborns are unavailable. Third, the overall cost of commercial grade clinical WGS/WES services is high. Although NGS technology costs have been dramatically reduced, they constitute a small proportion of the overall cost, which is dominated by clinical and reimbursement staffing and ancillary assay costs that do not decrease over time.
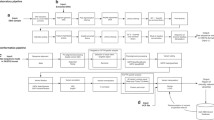
A targeted next-generation sequencing (TNGS) assay can cost-effectively address second-tier and diagnostic testing of newborns ( Figure 1a ) by selectively sequencing genomic regions of interest, typically coding exons, by enrichment in a physical DNA capture step ( Figure 1b ). TNGS can be re-purposed to also provide comprehensive coverage of elements such as introns. In many situations, the indicated symptoms can guide a focused investigation of specific disease genes (in silico gene filter; Figure 1a ). This has the advantages of a rapid test, lower cost of interpretation, and avoidance of delays encountered with serial single-gene testing and ethical concerns of genome-scale NGS (surrounding unrecognized pathologic variants or unanticipated findings).

Algorithm and workflow for next-generation sequencing (NGS)-based newborn confirmatory and diagnostic testing. (a) Exome (whole-exome sequencing, WES) and 126-gene panel (NBDx) analysis using in silico gene filters. A subset of the exome or targeted NGS panel can be interpreted utilizing a simple newborn-disease-specific in silico gene filter that includes diseases interrogated through NBS, or disorders that have a chance of early onset or presenting symptoms in the neonatal intensive care unit (NICU). This focused approach minimizes the problem of incidental findings and variants of unknown significance. Custom panels may be applied, for example, to identify sequence abnormalities in hearing loss genes, or genes associated with other common NICU phenotypes. Secondary validations will be required by Sanger sequencing or alternative technologies, in which there are exceptions, or for secondary confirmation of cis or trans heterozygotes. (b) Workflow for WES and NBDx. (1) DNA is isolated from a patient blood sample, including dried blood spot, commonly used for newborn screening. (2) Target DNA capture using Roche Nimblegen SeqCap EZ WES Library v2.0 and sequencing on the Illumina HiSeq 2000 or 2500 (3) Raw data sequence calling and alignment by Genome Analysis Toolkit v2.0 (4) Analysis and interpretation of variant calls by Omicia.
It is impractical for newborns, who have small total blood volumes, to routinely provide the 2 to 10 ml of whole blood typically requested for high-quality NGS services. Minimally invasive specimen types, such as DBS (wherein one spot is equivalent to 50 µl),12 are widely utilized for newborns, and if incorporated into the NGS workflow, will be more practical—avoiding the stringent specimen handling that is otherwise necessary and allowing accessibility in low-resource environments.
Time to results is critical for prompt treatment and management of life-threatening genetic disorders in newborns. The rapid diagnosis of phenylketonuria (OMIM 261600) from DBS,2,12 combined with early initiation of a low-phenylalanine diet and/or tetrahydrobiopterin therapy, demonstrated how NBS can prevent onset of symptoms, including profound mental retardation. Over the years, with technological advances, additional treatable or manageable metabolic disorders have been added to NBS programs.13,14,15,16,17,18
Second-tier DNA molecular analysis after NBS has been used for confirmation of common mutations for sickle cell disease (OMIM 141900),19 cystic fibrosis (OMIM 602421),20,21 and several other metabolic disorders,22,23,24 but these mutations may vary in frequency depending on population, limiting its utility. NGS-based second-tier testing has the advantage of improving performance of the primary biochemical NBS by reducing false positives (and parental anxiety), identifying de novo variants, and distinguishing genotypes associated with milder phenotypes (e.g., the mild R117H compared with the common pathological ΔF508 in cystic fibrosis).21 NGS second-tier DNA testing also lays the foundation for future primary DNA-based screening programs, especially for disorders such as cystinosis (OMIM 219800) that are not readily detectable via biochemistry. Recently, second-tier testing using amplicon NGS has been proposed for severe combined immunodeficiency disease (OMIM 300400) after primary screening of T-cell receptor excision circles from DBS.25,26
We undertook the development of fast-turnaround, minimally invasive, and cost-effective clinical sequencing and reporting in newborns, allowing a simplified testing menu that utilizes both WES and a more focused panel approach (NBDx), along with in silico gene filters ( Figure 1a and Table 1 ). In a retrospective proof-of-concept study, we determined the performance in the context of sequence variants associated with metabolic and other genetic disorders responsible for common phenotypes in the neonate.
Materials and Methods
Patient Samples
Validation specimens, unless stated otherwise, were obtained from patients with known causal mutations in the Amish and Mennonite populations examined at the Clinic for Special Children (CSC) in Strasburg, Pennsylvania. Specimens were collected under informed consent as part of diagnostic and research protocols approved by both the Lancaster General Hospital and the Western Institutional Review Boards. In this cohort, the disease-causing mutations were initially characterized by traditional Sanger DNA sequencing and were blinded for our NGS study. The clinic provides diagnosis and management of patients with inherited metabolic and genetic diseases within Amish and Mennonite populations. Mutations in the Amish and Mennonites are not unique, but they occur in higher frequencies than they do in the general population. The high incidence of disease and carrier cases can thus be used to validate the analytical test performance and genotype–phenotype concordance of new testing methodologies.
Sample Processing, Target Capture, and NGS
Briefly, isolated DNA was fragmented, barcoded with NGS library adapters, and incubated with oligonucleotide probes for DNA target capture, as outlined by the manufacturer (Roche Diagnostics, Indianapolis, IN), for all coding exons (SeqCap EZ Human Exome Library v2.0; 44-Mb target) or the NBDx targeted panel (SeqCap EZ Choice; up to 7-Mb target). Sequencing was performed with 2 × 75 bp HiSeq2500 rapid runs (Illumina, San Diego, CA). All NGS experiments were performed in research mode while keeping read depth and quality to mimic clinical grade metrics: >70% reads on target; >70× mean target base coverage; and >90% target bases covered >20×. An additional experiment used Nextera Rapid Capture (TruSight Inherited Disease; Illumina) for CYP21A2 testing on MiSeq.
NGS Analysis
Sequencing reads were aligned to hg19/GRCh37 using Burrows-Wheeler Aligner for short alignments,27 followed by Genome Analysis Toolkit v2.0 variant calling pipeline28,29 running on the Arvados platform (http://arvados.org). Opal 3.0 from Omicia (http://www.omicia.com) was used for variant annotation and analysis following guidelines of the American College of Medical Genetics.30
ClinVar Site Coverage Calculation
ClinVar sites (http://www.ncbi.nlm.nih.gov/clinvar/) were determined by intersecting the NBDx tiled regions with the ClinVar track in the UCSC Browser (http://genome.ucsc.edu/) and removing duplicates to give a total of 6,215 unique ClinVar sites.
Results
TNGS Workflow Test Using In Silico NBS Gene Filter and Rapid Turnaround
New NGS workflows are typically benchmarked against the traditional Sanger sequencing technology. CSC had previously identified more than 100 variants among the 120 different disorders identified at the clinic by Sanger sequencing, 32 of which were causal mutations for inborn errors of metabolism that are routinely screened by NBS. We used 10 of the CSC patient samples identified by such benchmark methods to optimize WES and in silico filtering for detection of the causal genetic variants.
The WES workflow was initially tested with two disease cases that are common in the Amish and Mennonite populations, propionic acidemia and maple syrup urine disease type 1A, to identify attributes of filtering regimens and causal variants ( Table 2 ). Simple filters for coverage, allele frequency, and pathogenicity reduced the number of variants in the WES samples from an average of 11,014 for exonic protein impact to 590. The in silico 126-gene NBS filter described in Table 1 reduced this to approximately four mutations, and the Sanger-validated causative homozygous mutations were easily identified. Thereafter, we undertook a blinded retrospective validation study using eight randomly selected samples from the same population to benchmark our results and demonstrate achievable turnaround times. The entire workflow from blood sample isolation through target capture, sequencing on a HiSeq 2500 in rapid run mode, informatics, and interpretation was parallel-processed within 105 hours for the eight WES samples ( Figure 1b ). Capture performance data indicated that, on average, 95% of the target bases were covered at 10× read depth or more and, of the total mapped reads, 73% were in WES target regions (Supplementary Table S1 online). Using the 126-gene NBS in silico filter, the correct disorder and mutation, as previously validated by Sanger sequencing, were quickly identified by TNGS in all eight samples. One subject was suspected to be a compound heterozygote for PAH ((c.782 G>A/c.284-286del) OMIM 261600), indicative of phenylketonuria. This subject also had a heterozygous carrier mutation in MCCC2 (OMIM 609014), which is commonly present in the Amish population. A similar situation was found in the subject with 11-β-hydroxylase deficiency, whereby a carrier of the c.646 G>A mutation responsible for adenosine deaminase deficiency was identified. This mutation is also known to segregate in the Amish population. All other samples were found to be homozygous for the common mutations known to occur in the Amish and Mennonite populations ( Table 2 ). Further, an alternate in silico gene filter representing 552 genes on the Illumina hereditary panel6 did not detect the mutations in IL7R and MTHFR (false-negative calls), which are genes that are not targeted in that panel.
Validation of DNA Isolation From Minimally Invasive DBS and Small-Volume Whole Blood for TNGS
We demonstrated robust and reproducible recovery of sufficient dsDNA from DBS for TNGS libraries (Supplementary Note S1 online), which, to our knowledge, has not been widely used except for research protocols in methylation assays.31,32 Using our methods, we see similarly high-quality TNGS performance of DNA isolated from DBS as compared with the standard 10 ml of whole blood and saliva (Supplementary Figure S1 online). With a control sample set, our protocols yielded ~450 ng double-stranded DNA (dsDNA) from one-half of a single saturated spot from the DBS card, representing 25 µl blood (as measured by the dsDNA-specific Qubit assay; Supplementary Table S2 online). The SeqCap EZ capture method used here requires 200 ng dsDNA, and an additional 10 to 20 ng for quality-control measurements. Recent methods of NGS library construction claim as little as 50 ng dsDNA for library construction (e.g., Nextera). Whole-genome amplification (WGA) could mitigate in cases of insufficient yield,33 and we have been successful in performing TNGS with DNA from DBS after WGA using Repli-G Ultrafast (Qiagen). In comparisons of matched samples, the addition of WGA resulted in approximately 5% lower target region covered at read depths 10× to 100× (Supplementary Figure S1b online), yet concordance remained near 100% across approximately 80 variants (data not shown).
Newborn-Specific Targeted Gene Panel (NBDx) Capture and NGS Performance
To measure NBDx gene panel performance, we tested 36 clinical samples that had mutations for metabolic diseases from the Amish and Mennonite populations ( Table 3 , Supplementary Table S3 online). Eight samples from this set were common with those of the WES analysis performed earlier. All samples were previously characterized by Sanger sequencing but were anonymized and thus interpreted in a blinded fashion regarding the disorder and mutation present. It was ultimately revealed that the samples had causative mutations in 18 separate disease-related genes. Eleven samples in the set showed 19 different mutations spanning across the glutaric acidemia type I gene, GCDH (arrows in Figure 2a ).

Newborn-specific targeted gene panel (NBDx) capture and sequencing performance. (a) Sensitivity plots for PAH and GCDH with matched whole-exome sequencing (WES)/NBDx pairs. Plot generation was based on sensitivity of 1, representing a coverage depth of ≥13 reads.35 Location of tiled probes for WES and NBDx across each RefSeq gene is shown. Mutations detected are indicated by arrows. (b) Fraction of ClinVar sites represented on WES and NBDx regions at different coverage depths. The percentage of ClinVar sites from the NBDx tiled regions with sequencing reads that reach increasing coverage depths, as determined using Samtools pileup (http://samtools.sourceforge.net). The graph includes data from eight matched samples captured for WES (red) and NBDx (blue).
Next, we compared NBDx for capture enrichment performance against WES. NBDx captures were processed at 20 samples per HiSeq2500 lane in rapid run mode, as compared with four samples for WES (Supplementary Table S1 online). The average reads on target were approximately twofold higher for NBDx compared with WES (151× vs. 88×) because of focused sequencing combined with a higher on-target specificity relative to WES (87% vs. 73%). Because read depth is a good predictor of variant detection (sensitivity), we used it to identify regions that are undercovered, i.e., less than 13 reads ( Figure 2a ). Sensitivity plots for GCDH and PAH across chromosomal positions were generated for WES or NBDx, as previously described by Meynert et al.34 As expected, compared with NBDx, WES had lower sensitivity because of lack of intronic probe coverage in PAH and GCDH.
The increased average sequencing depth in NBDx ensured that fewer targeted regions would fall below stringent variant calling thresholds.34,35 This was demonstrated in coverage of approximately 6,215 ClinVar sites common to both WES and NBDx tiled regions, which measured call coverage in regions of clinical relevance that can be monitored in every sample in real time ( Figure 2b ). Furthermore, while both NBDx and WES started with more than 99% at 1× coverage, disparities began to show at 10× coverage; by 100× coverage, NBDx maintained 80% ClinVar coverage, but WES significantly declined to 39%. At 10× coverage, NBDx achieved close to 99.8% coverage, and at 1× coverage it achieved 99.99% coverage. We also empirically determined, by pooling samples and by allele dilution of rare pathogenic variants (e.g., GCDH (c.1262 C>T)), that heterozygous calls up to one-sixth proportion were called (observed as 18 reads out of 120 total reads for this variant in NBDx; other data not shown).
To assess uniformity or relative abundance of different targeted regions, we compared base distribution coverage. We obtained good uniformity on NBDx data sets, but WES data showed significant skew toward low coverage, which is likely to reduce confidence on zygosity calls (Supplementary Figure S2a online). To assess reproducibility, we performed comparisons for coverage depth at variant positions across matched data set pairs resulting from independent sample preparation and sequencing. The analysis suggested that DBS, 25 µl whole blood and saliva produced a similar proportion of calls with a high agreement (Pearson correlation coefficient = 0.9) between replicates (Supplementary Figure S2b online). Another aspect of reproducibility we measured is the variability of coverage between runs in tiled regions. For 12 samples, we charted the portion of the targeted region with sufficient coverage to achieve 95% sensitivity for heterozygous calls (>13 reads). The maximum value per region was designated as 1. The tiled regions, for which at least one sample had a value less than 1, are shown in Supplementary Table S4 online. From comparisons across 4 to 20 unrelated TNGS samples and a simple statistic (Z-scoring), we detected highly variable regions such as homozygous intronic deletions in PCCB between exons 10 and 11.
NGS Genotype Call Concordance
To assess the genotype concordance, we compared our NGS genotype calls to a priori–generated Sanger sequencing calls from the 36 subjects at CSC. The variations ranged across a variety of mutation types, including nonsynonymous variations, indels, stop gained, and intronic/splice site variations ( Table 3 and Supplementary Table S3 online). Concordance of disease calls based on NGS genotypes was determined according to two scenarios. The first was fully blinded to the condition present and only the NGS variant data were used to classify the genotype and assignment to a disease, whereas in the second scenario a description of the clinical phenotype was available to optimize the genotype call. Two damaging heterozygotes variants in the same disease gene were preliminarily assumed to be in trans until confirmation could be obtained from the de-blinded data. In patients, phasing of such haplotypes would typically be performed through Sanger sequencing of parents after NGS.
Using NGS genotype calls, we were able to make preliminary disease calls in 27 out of 36 cases blindly (75%), suggesting difficulty of correctly classifying disease variants without clinical phenotype information. Complications (as noted in Table 3 ) included the following: (i) inability to distinguish causal variants from other mutations, either dominant or variants of unknown significance (VUS) with a predicted “damaging” classification; (ii) variant calling errors that were found on de-blinding for clinical phenotype, but, once corrected, these cases were processed through our standard filtering regimen (Supplementary Figure S3 online); (iii) no gene coverage (see CYP21A2 below); and (iv) compound heterozygotes with an intronic second mutation, which require additional phenotype information. Clinical description plus a heterozygous damaging mutation in a disease-related gene enabled efficient intronic analysis within the same gene. Samples 9226 and 14691 had a combination of intronic mutations and heterozygosity in multiple genes.
A re-analysis with clinical summaries confirmed correct identification of mutations in seven additional disease or carrier cases, whereas two disease cases remained undetermined (ID 21901 and 27244) because the disease gene CYP21A2 was not targeted because of high pseudogene homology; however, false-positive calls were not made on these samples. A separate capture using the Illumina hereditary panel,6 that included CYP21A2, also failed to map the correct call. Two of the seven samples were carrier-status only (ID 23275 and 30221). Thus, with clinical phenotype, correct classification was obtained for 32 out of 34 disease cases (94.12%; 95% confidence interval, 80.29%–99.11%).
Discussion
Identifying genetic disorders in newborns typically uses a tiered approach. Asymptomatic newborns who are identified as being at risk for disorders by NBS receive confirmation with second-tier testing (biochemical or genetic) on a repeat sample obtained from the patient in question. However, the genetic etiology, delayed onset, and/or “milder phenotype” are probably missed. Symptomatic newborns, such as those admitted to a NICU, undergo an initial clinical assessment and sequential diagnostic testing to “rule out” causation; these require nomination based on history or clinical opinions, thus limiting the diagnostic rate and efficiency.10,11 Because blood draws are also of concern in newborns, it makes practical sense to utilize a single multigene sequencing panel to minimize sequential analysis and avoid delayed diagnosis.
The approach of using gene panels and in silico filters provides a systematic parallel or iterative review of symptom(s) and diseases from a molecular standpoint by providing information on the exact genes, their variant(s), and associated future risks (for family planning because of parental carrier status). The risks of potential harm attributable to variants of unknown significance, incidental findings, or false positives on NBDx are several orders of magnitude lower than in WGS and are not much different from existing concerns regarding current NBS algorithms or single-gene testing. It is important to realize that the burden of disease mutations and their combinations on phenotype or effect of cumulative mutations in genetic pathways that may act synergistically is not clearly monitored by NBS or single-gene sequencing for newborn diseases. Even for the limited in silico filter size of 126 genes and 36 cases studied here, we found 19 incidental carrier mutation that were previously described in the Amish and Mennonite populations ( Table 3 and Supplementary Table S4 online), indicating that such information should help in identifying subclinical traits and reproductive planning.
In the context of neonatal care, genomic tests like NBDx and WES can, as part of a testing menu, precisely inform in one test what the prenatal tests, ultrasounds, amniocentesis, and NBS test sometimes cannot. WES as a first-tier screening approach in newborns has not been recommended by the recent American College of Medical Genetics policy statement.36 However, as a secondary screen and guided by phenotype, it is consistent with the policy statement. Both NBDx and WES still require ethical considerations (e.g., to determine psychological impact of being found to be a carrier). Diagnosis can be helpful, even when no therapies are available, and allows parents of affected children to be informed about their care up-front and receive genetic counseling regarding the risk for future pregnancies.6
The developed comprehensive rapid test workflow for second-tier NBS testing and high-risk diagnosis of newborns for multiple genetic disorders is approaching the 2- to 3-day turnaround necessary for newborns to avoid medical sequelae. The test can currently be parallel-processed for 8 to 20 samples per lane and completed in 105 hours (approximately 4.5 days); and several approaches to reduce turnaround time show promise, such as alternate library preparation and reduced hybridization time. In cases in which mutations are suspected to be in trans, additional follow-up testing will be required. A significant milestone we have demonstrated here is the minimally invasive isolation of high-quality dsDNA from DBS and small blood volumes (25–50 µl) in sufficient amounts for TNGS. Adoption of DBS-based NGS testing may significantly reduce the burden of using more expensive lavender (purple) top tubes for blood collection, which add to special handling, shipping, and storage costs. Moving an NGS test to DBS enables widespread utility using centralized NGS testing facilities. When available, cord blood could be used as an alternative minimally invasive biological specimen source for TNGS, or dried on a card, similar to current DBS, for simplified transport.
When disease heterogeneity or multigene diseases are encountered during the newborn period (e.g., phenylketonuria, severe combined immunodeficiency disease, maple syrup urine disease, propionic acidemia, glutaric acidemia), a TNGS assay covering approximately 100 to 300 disease genes is as cost-effective as Sanger sequencing test(s) for quickly confirming or “ruling out” clinical suspicion or false-positive results.26,37 The cost of NBDx (Supplementary Note S2 online) is significantly less than that of WES, and both tests are expected to be similar in price range to diagnostic tests currently on the market and therefore should enable replacement of single-gene tests, as justified by delays and increased patient-management costs.6,10,11
We established performance benchmarks supporting direct clinical use similar to WGS newborn/pediatric testing of Mendelian diseases.6 In the NICU setting, either WES or NBDx adapted for minimal invasive sample size or rapid turnaround may assist in detecting mutations and diagnosing the critically ill, some of whom may have metabolic decompensation at birth. Even after NBS, cases of cystic fibrosis and metabolic conditions are routinely missed (false negatives) because of various causes, including biochemical cutoffs. This suggests NGS-based testing has the potential to add to sensitivity; however, critically ill newborn populations would need to be surveyed. We did not extend our studies to WGS because we see several performance challenges to clinical adoption. A recent study noted that 10 to 19% of inherited disease genes were not covered by WGS at accepted standards for single-nucleotide variant discovery.38 By contrast, in real time we noted 99.8% analytical sensitivity of ClinVar coverage in NBDx panels because of higher base coverage than the WES panel. Because deletion analysis and deep intronic or promoter variations are typically not covered in WES, it is likely to increase false negatives, as we have observed across PCCB intronic regions. We have also detected exon deletion in one maple syrup urine disease case (data not shown).
Our ability to pinpoint the clinical phenotype of an individual on the basis of “genotype” alone is still in its infancy; in our case, only 27 of 36 NBS disease cases were classified correctly without phenotype information. It is typically assumed that, at least for monogenic disorders, the genotype–phenotype relationship would be simple. However, many instances abound when, despite a classic disease-causing mutation, the phenotype is absent. Phenotypic information as part of NBS or clinical diagnosis can improve genotype call. Thus, with the clinical phenotype description, single-nucleotide variations in exons, introns (up to 30 bp away from an exon), and indels were correctly detected in 32 of 34 Amish or Mennonite disease cases and two carrier cases. It is foreseeable that with phenotypic information, a heuristic variant- and disease-calling pipeline can be built and automated.39
We also observed that compound heterozygous conditions are often not callable from NGS alone because current technologies cannot differentiate between cis or trans phasing. Of all 36 cases, identification of the disease-causing mutation was only missed in two cases (false negatives), with both being coding mutations in CYP21A2, which has a 98% homology to its pseudogene (CYP21A1) and frequently undergoes gene-conversion events. Thus, this region is not callable using hybridization-based assays combined with short-read NGS sequencing.40
Two compelling forces are expected to drive adoption of genetic testing in newborns with symptoms. First is the need for rapid, minimally invasive diagnosis to treat and minimize adverse outcomes. Second is the financial incentive to shorten length of stay and reduce overall patient-management costs associated with delayed or inaccurate diagnosis. This study demonstrates that turnaround and sample requirements for newborn genetic cases are achievable using TNGS, and that combining genetic etiology (via TNGS) with phenotype will help us arrive at a comprehensive clinical understanding promptly. Larger prospective studies using newborns should reveal more regarding clinical utility, diagnostic rates, and added value over the current standard of care.
Disclosure
A.B. declares he is an employee, founder, and stockholder in Parabase Genomics. T.S. declares she is an employee of Parabase Genomics and has stock ownership in Parabase Genomics. S.K.W. is a consultant for Parabase Genomics. M.G.R. declares he is an employee, founder, and stockholder in Omicia Inc. E.P., K.S., and H.M. declare no conflict of interest other than compensation received for providing samples and supporting data analysis. E.W.N. and R.B.P. declare they are stockholders and scientific advisers to Parabase Genomics.
References
Christianson A, Howson CP, Modell B . Global Report on Birth Defects: The Hidden Toll of Dying and Disabled Children. March of Dimes Birth Defects Foundation: White Plains, NY, 2006.
Landau YE, Lichter-Konecki U, Levy HL . Genomics in newborn screening. J Pediatr 2014;164:14–19.
The President’s Council on Bioethics. http://www.bioethics.gov/reports/newborn_screening/index.html. 2008. Accessed 28 August 2014.
Costa T, Scriver CR, Childs B . The effect of Mendelian disease on human health: a measurement. Am J Med Genet 1985;21:231–242.
Kumar P, Radhakrishnan J, Chowdhary MA, Giampietro PF . Prevalence and patterns of presentation of genetic disorders in a pediatric emergency department. Mayo Clin Proc 2001;76:777–783.
Saunders CJ, Miller NA, Soden SE, et al. Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci Transl Med 2012;4:154ra135.
Kaiser J . Genomics. Researchers to explore promise, risks of sequencing newborns’ DNA. Science 2013;341:1163.
Ng SB, Turner EH, Robertson PD, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 2009;461:272–276.
de Ligt J, Willemsen MH, van Bon BW, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 2012;367:1921–1929.
Shashi V, McConkie-Rosell A, Rosell B, et al. The utility of the traditional medical genetics diagnostic evaluation in the context of next-generation sequencing for undiagnosed genetic disorders. Genet Med 2014;16:176–182.
Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 2013;369:1502–1511.
Guthrie R, Susi A . A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics 1963;32:338–343.
Dussault JH, Laberge C . [Thyroxine (T4) determination by radioimmunological method in dried blood eluate: new diagnostic method of neonatal hypothyroidism?]. Union Med Can 1973;102:2062–2064.
Crossley JR, Elliott RB, Smith PA . Dried-blood spot screening for cystic fibrosis in the newborn. Lancet 1979;1:472–474.
Naylor EW . Recent developments in neonatal screening. Semin Perinatol 1985;9:232–249.
Pang SY, Wallace MA, Hofman L, et al. Worldwide experience in newborn screening for classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Pediatrics 1988;81:866–874.
Naylor EW, Chace DH . Automated tandem mass spectrometry for mass newborn screening for disorders in fatty acid, organic acid, and amino acid metabolism. J Child Neurol 1999;14(suppl 1):S4–S8.
Chace DH, Kalas TA, Naylor EW . The application of tandem mass spectrometry to neonatal screening for inherited disorders of intermediary metabolism. Annu Rev Genomics Hum Genet 2002;3:17–45.
Herrmann MG, Dobrowolski SF, Wittwer CT . Rapid beta-globin genotyping by multiplexing probe melting temperature and color. Clin Chem 2000;46:425–428.
Spence WC, Paulus-Thomas J, Orenstein DM, Naylor EW . Neonatal screening for cystic fibrosis: addition of molecular diagnostics to increase specificity. Biochem Med Metab Biol 1993;49:200–211.
Comeau AM, Parad RB, Dorkin HL, et al. Population-based newborn screening for genetic disorders when multiple mutation DNA testing is incorporated: a cystic fibrosis newborn screening model demonstrating increased sensitivity but more carrier detections. Pediatrics 2004;113:1573–1581.
Andresen BS, Dobrowolski SF, O’Reilly L, et al. Medium-chain acyl-CoA dehydrogenase (MCAD) mutations identified by MS/MS-based prospective screening of newborns differ from those observed in patients with clinical symptoms: identification and characterization of a new, prevalent mutation that results in mild MCAD deficiency. Am J Hum Genet 2001;68:1408–1418.
Naylor EW, Hoffman EP, Paulus-Thomas J, et al. Neonatal screening for Duchenne muscular dystrophy: reconsideration based on molecular diagnosis and potential therapeutics. Screening 1992;1:99–113.
Dobrowolski SF, Banas RA, Suzow JG, Berkley M, Naylor EW . Analysis of common mutations in the galactose-1-phosphate uridyl transferase gene: new assays to increase the sensitivity and specificity of newborn screening for galactosemia. J Mol Diagn 2003;5:42–47.
Puck JM . Neonatal screening for severe combined immune deficiency. Curr Opin Allergy Clin Immunol 2007;7:522–527.
Saavedra-Matiz CA, Isabelle JT, Biski CK, et al. Cost-effective and scalable DNA extraction method from dried blood spots. Clin Chem 2013;59:1045–1051.
Li H, Durbin R . Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–1760.
McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–1303.
DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43:491–498.
Richards CS, Bale S, Bellissimo DB, et al.; Molecular Subcommittee of the ACMG Laboratory Quality Assurance Committee. ACMG recommendations for standards for interpretation and reporting of sequence variations: revisions 2007. Genet Med 2008;10:294–300.
Beyan H, Down TA, Ramagopalan SV, et al. Guthrie card methylomics identifies temporally stable epialleles that are present at birth in humans. Genome Res 2012;22:2138–2145.
Aberg KA, Xie LY, Nerella S, Copeland WE, Costello EJ, van den Oord EJ . High quality methylome-wide investigations through next-generation sequencing of DNA from a single archived dry blood spot. Epigenetics 2013;8:542–547.
Hollegaard MV, Grauholm J, Nielsen R, Grove J, Mandrup S, Hougaard DM . Archived neonatal dried blood spot samples can be used for accurate whole genome and exome-targeted next-generation sequencing. Mol Genet Metab 2013;110:65–72.
Meynert AM, Bicknell LS, Hurles ME, Jackson AP, Taylor MS . Quantifying single nucleotide variant detection sensitivity in exome sequencing. BMC Bioinformatics 2013;14:195.
Ajay SS, Parker SC, Abaan HO, Fajardo KV, Margulies EH . Accurate and comprehensive sequencing of personal genomes. Genome Res 2011;21:1498–1505.
Green RC, Berg JS, Grody WW, et al.; American College of Medical Genetics and Genomics. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med 2013;15:565–574.
Prach L, Koepke R, Kharrazi M, et al.; California Cystic Fibrosis Newborn Screening Consortium. Novel CFTR variants identified during the first 3 years of cystic fibrosis newborn screening in California. J Mol Diagn 2013;15:710–722.
Dewey FE, Grove ME, Pan C, et al. Clinical interpretation and implications of whole-genome sequencing. JAMA 2014;311:1035–1045.
Singleton MV, Guthery SL, Voelkerding KV, et al. Phevor combines multiple biomedical ontologies for accurate identification of disease-causing alleles in single individuals and small nuclear families. Am J Hum Genet 2014;94:599–610.
Mueller PW, Lyons J, Kerr G, Haase CP, Isett RB . Standard enrichment methods for targeted next-generation sequencing in high-repeat genomic regions. Genet Med 2013;15:910–911.
Acknowledgements
This work has been funded by the phase I SBIR NIH grants from NIDCD (1R43DC013012-01) and NICHD (1R43HD076544-01) to A.B. and E.W.N. (Parabase Genomics principal investigators), as well as generous financial support of early-stage investors in Parabase Genomics. We thank Yael Katan-Khaykovich, Angelica Laing, and Robert Liebman for DNA preparation and assay development. We thank David B. Gordon of MIT/Broad Institute, Adam Berrey and Ward Vandewege of Curoverse, and Edward Kiruluta of Omicia for their valuable support and advice. We thank Jordan Lerner-Ellis at University of Toronto for early data generation on the HiSeq2000 platform, Irina Tikhonova and colleagues for sequencing support at Yale Sequencing Center, and several faculty members in the Biology Department at University of Massachusetts, Boston, Massachusetts, for access to MiSeq platform, and Symmetric Computing Inc., Boston, Massachusetts, for access to a departmental supercomputer. All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Declaration of Helsinki of 1975, as revised in 2000. Informed consent was obtained from all patients included in the study.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Information
(DOC 873 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Bhattacharjee, A., Sokolsky, T., Wyman, S. et al. Development of DNA Confirmatory and High-Risk Diagnostic Testing for Newborns Using Targeted Next-Generation DNA Sequencing. Genet Med 17, 337–347 (2015). https://doi.org/10.1038/gim.2014.117
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2014.117
Keywords
This article is cited by
-
A pediatric perspective on genomics and prevention in the twenty-first century
Pediatric Research (2020)
-
Using dried blood spot samples from a trio for linked-read whole-exome sequencing
European Journal of Human Genetics (2019)
-
The BabySeq project: implementing genomic sequencing in newborns
BMC Pediatrics (2018)
-
Genomic newborn screening: public health policy considerations and recommendations
BMC Medical Genomics (2017)
-
Array comparative genomic hybridization and genomic sequencing in the diagnostics of the causes of congenital anomalies
Journal of Applied Genetics (2017)
