
Abstract
Purpose:
Sapropterin is an oral synthetic formulation of tetrahydrobiopterin prescribed as adjunctive therapy for phenylketonuria. The efficacy of sapropterin in reducing blood phenylalanine levels has been demonstrated in clinical studies of individuals with phenylketonuria older than 4 years of age. Its effect on neurocognitive functioning in younger children has not been examined.
Methods:
A 2-year interim analysis of blood phenylalanine levels, prescribed dietary phenylalanine intake, and neurocognitive functioning was performed in children who started receiving sapropterin at 0–6 years of age and responded with a ≥30% mean blood phenylalanine reduction. Children were evaluated at baseline and 2-year follow-up.
Results:
Sapropterin had a favorable safety profile and lowered blood phenylalanine levels with increased prescribed dietary phenylalanine intakes. Mean full-scale intelligence quotient was 103 ± 12 at baseline and 104 ± 10 at 2-year follow-up (P = 0.50, paired t-test, n = 25). For children younger than 30 months of age, the cognitive composite score from the Bayley Scales of Infant and Toddler Development, Third Edition, remained within the average range.
Conclusion:
Sapropterin had a favorable safety profile, was effective in lowering blood phenylalanine levels while clinically requiring dietary adjustment, resulting in increased phenylalanine intake, and preserved neurocognitive performance in children who started therapy between 0 and 6 years of age.
Genet Med 17 5, 365–373.
Similar content being viewed by others


Introduction
Phenylketonuria (PKU) is an autosomal recessive disorder caused by deficiency of phenylalanine hydroxylase, resulting in impaired conversion of the essential amino acid phenylalanine (Phe) to tyrosine.1 The accumulation of Phe in the blood (hyperphenylalaninemia) and brain can cause intellectual disability, seizures, behavioral difficulties, and other symptoms.2,3 With neonatal screening and early initiation of treatment, the most severe neurodevelopmental consequences of PKU can be successfully prevented.4,5,6
Treatment of PKU involves a low-phenylalanine diet to reduce blood Phe levels and to prevent the development of intellectual disability.7 The low-Phe diet is highly restrictive because Phe is an amino acid found in all proteins. The diet is socially burdensome for many individuals and may hinder physical growth.8 It requires avoidance of meat, fish, dairy products, nuts, beans, and other protein-containing foods. Only measured amounts of Phe-containing intact protein are allowed from fruits, vegetables, breads, and other starchy foods.9 Synthetic Phe-free medical foods and/or supplemental formula provide other essential amino acids and tyrosine, but many have a strong taste and odor that many individuals find unpleasant.10
Early and continuous dietary therapy prevents the more severe neurological and intellectual dysfunction associated with classic PKU, but more subtle deficits in intelligence, academic achievement, and executive functioning can occur even in individuals with PKU who are treated early.11,12,13 Despite functioning within the average range on tests of intelligence, children and adults with PKU have a lower mean intelligence quotient (IQ) than the general population.12,14,15 Deficits affecting language skills, visual-spatial abilities, processing speed, verbal comprehension, and perceptual reasoning skills have also been reported.13,16,17,18,19,20 A meta-analysis of Phe levels and IQ in 40 studies confirmed a significant negative correlation between blood Phe levels and IQ in individuals with PKU who were treated early.21 Significant proportional correlations were present between Phe level and IQ during the critical period from 0 to 12 years of age, in which each 100-μmol/l increase in Phe was associated with a reduction of 1.3 to 3.1 IQ points. Similarly, significant correlations were observed between IQ and mean lifetime Phe level for patients treated early, in which each 100-μmol/l increase in Phe predicted a reduction of 1.9 to 4.1 IQ points.21
Tetrahydrobiopterin (BH4) is an essential cofactor of phenylalanine hydroxylase. Sapropterin dihydrochloride is an oral synthetic formulation of BH4 available as an adjunctive therapy to a Phe-restricted diet to reduce elevated blood Phe levels in patients with hyperphenylalaninemia attributable to BH4-responsive PKU. The efficacy of sapropterin in reducing blood Phe levels has been demonstrated in clinical studies of children older than 4 years of age and adults with PKU.22,23,24,25 In a recent study of 15 children younger than age 4 years with PKU, sapropterin improved blood Phe control while clinically requiring dietary Phe adjustments, allowing increased intact protein intakes; however, the impact of sapropterin on neurocognitive development was not assessed.26 A retrospective analysis of children and adults with BH4-responsive PKU demonstrated that the improved metabolic control and dietary adjustments associated with sapropterin therapy were maintained long term (up to 12 years).27
This study was conducted to prospectively evaluate the long-term efficacy of sapropterin in preserving neurocognitive function in children with PKU who began sapropterin treatment between 0 and 6 years of age.
Materials and Methods
Patient selection
Patients eligible for this study were children aged 0 to 6 years at the time of screening with a diagnosis of PKU/hyperphenylalaninemia and at least two blood Phe concentrations ≥360 μmol/l (6 mg/dl) taken at least 3 days apart. Documented compliance with local treatment standards was required before study enrollment. A parent or guardian had to sign consent and be able to comply with all study procedures, including adherence to a prescribed Phe-restricted diet designed to maintain blood Phe concentrations within the recommended ranges established at the study site.
Children were not eligible for the study if they had hypersensitivity to sapropterin or its excipients, a diagnosis of primary BH4 deficiency, history of organ transplantation, a serious neuropsychiatric illness (e.g., major depression) not currently medically under control, or a concurrent disease or condition that would interfere with study participation or safety (e.g., seizure disorder, insulin-dependent diabetes, oral steroid–dependent asthma, other condition requiring oral or parenteral corticosteroid administration). Children using the following medications were excluded from the study (per the sapropterin prescribing information caution about coadministration with these drugs): phosphodiesterase type 5 inhibitors, methotrexate, or medications that inhibit folate metabolism. Children who had used sapropterin or any investigational product (drug or medical device) within 30 days before screening were also excluded.
This study was conducted in compliance with the International Conference on Harmonisation Guideline for Good Clinical Practice and the ethical principles for research on human subjects in the Declaration of Helsinki. The study protocol was approved by the institutional review board, ethics committee, or research ethics board of each participating institution or a central institutional review board. Written informed consent was obtained from each subject’s parent or legal guardian before study enrollment.
Study design
The PKU-015 study is an ongoing, multicenter, open-label study evaluating the safety of sapropterin and its effect on maintaining neurocognitive function, blood Phe concentration, and growth in children with PKU who were 0 to 6 years old when initiating treatment. The study will continue until ~45 subjects complete 7 years of treatment. This article reports the interim analysis of data for a subset of patients who responded to sapropterin therapy and had completed at least 2 years in the study as of June 2012, as well as three patients who terminated early from the study before their 2-year study visit (see study flow diagram in Figure 1 ).

Study flow diagram with patient disposition. AE, adverse events; Phe, phenylalanine; PI, principal investigator.
Study participants were required to adhere to a prescribed Phe-restricted diet to maintain blood Phe concentrations within recommended ranges (<360 μmol/l), with a goal of maintaining blood Phe concentration between 120 and 240 μmol/l. Study participants and their parent(s) or guardian(s) met with a study dietitian at each visit to review dietary Phe intake using a 3-day food record.
The first 4 weeks of the study consisted of an evaluation of responsiveness to sapropterin at 20 mg/kg daily. Participants returned to the clinic weekly for safety assessments and measurement of blood Phe concentration. After completion of the first 4 weeks of the study, only subjects who were responsive to sapropterin continued the study. Sapropterin responsiveness was defined as a ≥30% average reduction in blood Phe concentration from baseline calculated from the average of phenylalanine levels at weeks 1, 2, 3, and 4. Sapropterin-responsive subjects received a baseline neurocognitive assessment within 6 weeks of confirmation of sapropterin responsiveness.
Subjects who responded to sapropterin and attained a score of ≥80 on the infant developmental test or an IQ ≥80 were eligible to continue in the study, which included a 6-month safety and efficacy evaluation followed by a long-term neurocognitive evaluation for 7 years of follow-up. Study participants continued to receive single oral doses of 20 mg/kg sapropterin daily. Dose reduction was permitted after week 5 in children who did not tolerate the 20 mg/kg/day dose. Study visits occurred monthly up to 1 year and every 6 months thereafter through year 7. Interim assessments were conducted by telephone every 3 months to assess weight, adverse events (AEs), and concomitant medications.
For all phases of the study, recommended dietary Phe intake remained constant at the level prescribed before study enrollment, except if blood Phe concentration dropped below 120 μmol/l. Then, a gradual increase of approximately 5 to 20 mg/kg of daily dietary Phe supplement was permitted at the metabolic physician’s discretion. A reduction of prescribed dietary Phe by approximately 5 to 20 mg/kg (or per the standard at each treatment center) was permitted if a patient’s blood Phe level increased higher than 240 μmol/l.
Study drug
Sapropterin dihydrochloride (Kuvan; BioMarin Pharmaceutical, Novato, CA) is an orally active, synthetic version of the 6R-isomer of BH4, a naturally occurring cofactor of phenylalanine hydroxylase that stimulates activity of the residual phenylalanine hydroxylase enzyme to metabolize Phe into tyrosine.
Efficacy variables
The primary objective of this study was to determine the long-term efficacy of sapropterin in preserving neurocognitive function in children with PKU when treatment is started at 0 to 6 years of age. Neurocognitive testing was performed in all sapropterin-responsive patients within 6 weeks of determination of responsiveness (i.e., baseline for the neurocognitive study). The Bayley Scales of Infant and Toddler Development, Third Edition (Bayley-III), were administered every 6 months to children aged 0 months to <30 months. The Wechsler Preschool and Primary Scale of Intelligence, Third Edition (WPPSI-III), was administered annually for children aged ≥30 months to <7 years. The Wechsler Intelligence Scale for Children, Fourth Edition (WISC-IV), was administered every 2 years to children 7 years of age or older.
Secondary efficacy variables included long-term safety of sapropterin, efficacy of 6 months of sapropterin treatment in controlling blood Phe concentration, and effect of sapropterin on growth parameters (i.e., change from baseline). Plasma blood Phe levels were measured at months 1, 3, 6, 12, 18, and 24 by local laboratories using local protocols. Height and head circumference were measured at screening, week 0, week 4, month 3, month 6, and every 6 months thereafter. Weight was measured at every visit.
Safety variables
AEs, including clinically significant laboratory findings and new or exacerbated symptoms or diseases, were recorded at each visit and classified by severity (mild, moderate, or severe) and likelihood of relationship to the study drug (not related, possibly related, or probably related). Serious AEs were defined per the International Conference on Harmonisation as events that led to or prompted treatment, including death, dismemberment, birth defect, hospitalization, and prolongation of hospitalization, or were otherwise clinically significant in the opinion of the investigator.
Statistical analysis
Full-Scale Intelligence Quotient (FSIQ) constituted the primary efficacy end point. The mean FSIQ from the WPPSI-III or WISC-IV at baseline was compared with mean FSIQ on these tests at the 2-year follow-up assessment using a paired t-test. To account for patients moving from WPPSI-III assessment to WISC-IV assessment as they aged, data were included in a combined analysis, stratified by testing sequence used (WPPSI-III/WPPSI-III, WPPSI-III/WISC-IV, or WISC-IV/WISC-IV). Data from the Bayley-III assessment were analyzed separately from WPPSI-III and WISC-IV and were used to ensure the safety of sapropterin for very young children in terms of early development. Absolute mean changes in blood Phe levels and prescribed dietary Phe from baseline to each follow-up time point were compared with t-tests. Changes in height, weight, and head circumference from baseline to 2 years were analyzed using a longitudinal model. The incidence of treatment-emergent AEs was summarized by severity and relationship to the study drug.
Results
Sample population
Of 95 children enrolled in the study, 71 were considered sapropterin responders and 65 continued to the 6-month safety and efficacy evaluation ( Figure 1 ). Six of the sapropterin responders did not continue in the study for a variety of reasons (noncompliance with treatment, AEs, error at the clinical site, and decision of the principal investigator) ( Figure 1 ). Of the 65 sapropterin responders who participated in the 6-month safety and efficacy evaluation, 63 continued beyond 6 months and were enrolled in the long-term evaluation of neurocognitive function. A total of 55 patients, enrolled at 11 genetics clinics in the United States and Canada, had data available for analysis of 2-year outcomes. Baseline and safety data for the three subjects who discontinued study participation before the 2-year follow-up evaluation were included in the 2-year analysis ( Figure 1 ).
Baseline characteristics are shown in Table 1 . The average age at enrollment was 3.1 ± 2.2 years. Two-thirds of participants were female, and most participants were white. The mean blood Phe concentration at baseline was 331 ± 138 μmol/l.
Sapropterin exposure and responsiveness
Among the 95 patients tested for sapropterin responsiveness, 75% (71 of 95) were either “per-protocol” sapropterin responders (i.e., ≥30% mean reduction of blood Phe concentration; n = 63) or “clinical” sapropterin responders (i.e., <30% mean blood Phe reduction but maintained blood Phe in the advised range, e.g., 120–360 μmol/l, despite increased dietary Phe intake; n = 8). Age distributions among sapropterin responders were: 85% (11/13) of children ages 0 to less than 1 year; 65% (24/37) of 1- to 2-year-olds; 81% (22/27) of 3- to 4-year-olds; and 78% (14/18) of 5- to 6-years-olds. Of the 55 children in the 2-year analysis, 48 were per-protocol sapropterin responders and 7 were clinical sapropterin responders.
Dosing compliance was 98–99% for all age groups in analyses based on completion of study drug regimen, days of correct dosage taken, and actual days on study drug. The average daily dose of sapropterin received by weight (mean, 20.3 mg/kg) varied little across age groups, indicating that most children were maintained on the initial dose of 20 mg/kg per day.
Phenylalanine control
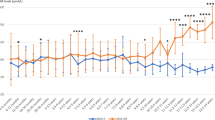
Mean blood Phe concentrations in the sapropterin-responder population declined markedly in all children in each age group from baseline to the week 4 visit, then increased to levels still below baseline by month 3, with the exception of 3- to 4-year-old children, in whom blood Phe increased to the baseline level ( Figure 2a ). Prescribed dietary Phe increased from baseline to 2-year follow-up in all age groups, both in absolute terms (mg/day) and when adjusted for body weight (mg/kg/day) ( Figure 2b , c ).

Mean blood phenylalanine (Phe) and prescribed dietary Phe from baseline to 2 years. (a) Phe levels were measured in children with phenylketonuria (PKU) at baseline and at the indicated time after starting therapy with sapropterin. (b,c) Prescribed dietary Phe in children with PKU at baseline and at the indicated time after starting therapy with sapropterin expressed in mg/kg of body weight (b) or total mg per day (c). Points are the mean ± SE at each time point, with the overall group including all patients. *P < 0.05 as compared with baseline using analysis of variance.
Phe levels were ≤240 μmol/l in 18/55 (33%) children at baseline, 42/53 (79%) at 1 week, 33/48 (69%) at 2 months, 35/52 (67%) at 6 months, 33/52 (63%) at 12 months, and 32/50 (64%) at 24 months. Phe concentrations were ≤360 μmol/l in 36/55 (65%) patients at baseline, 50/53 (94%) at 1 week, 40/48 (83%) at 2 months, 43/52 (83%) at 6 months, 44/52 (85%) at 12 months, and 42/50 (84%) at 24 months.
Development and neurocognitive function
Baseline neurocognitive testing indicated that participants had average FSIQ not significantly different from the population norm of 100. Among the 25 children for whom baseline and 2-year WPPSI-III and WISC-IV scores were available, the mean FSIQ was 103 ± 12 at baseline and 104 ± 10 at 2-year follow-up, with no statistically significant changes over time (P = 0.50, paired t-test). Mean scores on the Bayley-III cognitive composite index from baseline to 2 years were also maintained within the normative range of 100 ± 15, with no significant changes over time. After starting sapropterin therapy, no infant or toddler received a score less than 85 on the Bayley-III cognitive composite index, indicating that no child appeared to be at risk for developmental delay during the study.
AEs
Table 2 presents nonserious AEs classified as possibly or probably related to sapropterin and occurring in >5% of individuals with PKU. These included abdominal pain, diarrhea, vomiting, infections of the ear and upper respiratory tract, nasal congestion, and headache. Six serious AEs (also shown in Table 2 ) were reported in five (9%) subjects. None of the serious AEs, which included constipation, croup, pneumonia, injury, anesthesia complication, and seizure, was deemed by the investigator to be related to sapropterin.
Growth assessments
Mean z-scores for height, weight, and head circumference are shown in Figure 3 . Baseline z-scores for all three growth parameters (height 0.4 ± 0.9, weight 0.4 ± 0.8, and head circumference 0.3 ± 1.0) were slightly above the 50th percentile for the 2000 Centers for Disease Control and Prevention reference values and were maintained throughout 2 years of follow-up in children taking sapropterin, with no statistically significant changes from baseline.

Growth assessments in patients with phenylketonuria. (a) Height; (b) weight; (c) head circumference. z-Score, the deviation of the value for an individual from the mean value of the reference population divided by the standard deviation for the reference population based on Centers for Disease Control and Prevention reference values established in 2000.
Discussion
Sapropterin dihydrochloride, a synthetic formulation of BH4, can reduce elevated blood Phe levels in individuals with hyperphenylalaninemia, but limited data are available for young children. In this study, 75% of children with PKU aged 0 to 6 years responded to sapropterin therapy ( Figure 1 ), with 66% meeting the ≥30% reduction in mean blood Phe levels. This proportion of sapropterin responders was higher than in previous studies. The proportions of sapropterin responders among older individuals with PKU taking sapropterin were 56% (50/89) in the study by Trefz and colleagues25 and 44% (18/41) in the study by Levy et al.,24 and both studies used ≥30% mean blood Phe reduction as the responsiveness criterion. The reason for the higher response rate in our study may reflect better-controlled dietary Phe intake in younger children, for whom the parents (rather than the patient) prepare and administer medical foods. Alternatively, our response rate may represent a bias of ascertainment, with preferential selection of patients more likely to respond (although all eligible patients were enrolled).
Mean blood Phe concentration decreased after initiation of sapropterin therapy and was maintained at ≤360 μmol/l in 84% of subjects and at ≤240 μmol/l in 64% of subjects at 2-year follow-up, despite increases in mean prescribed dietary Phe ( Figure 2 ). There was an increase in blood Phe levels after week 4 of therapy, reflecting an increase in prescribed dietary Phe (allowed by protocol starting at the week 5 visit to maintain blood Phe in the targeted range). Previous studies have indicated that sapropterin improves blood Phe control while clinically requiring dietary Phe adjustments, leading to less restrictive diets and, in some cases, discontinuation of Phe-free medical food.25,26,28,29 The current study confirms and extends this observation to younger children who have a higher growth velocity and, consequently, a relatively higher requirement for phenylalanine.
Neurocognitive outcomes
Young children with PKU treated with sapropterin had average IQ scores at baseline. Maintenance of metabolic control with a combination of dietary therapy and sapropterin preserved IQ throughout 2 years of therapy. Final analyses of follow-up assessments after 7 years of sapropterin treatment will provide additional information about the longer-term efficacy of early treatment with sapropterin therapy in addition to Phe-restricted dietary regimens in very young children.
Safety and tolerability
Sapropterin had a favorable safety profile and was well tolerated, based on dose adherence and absence of serious AEs, consistent with previous studies of sapropterin in subjects older than 4 and 8 years of age.23,24,25,30 The reported AEs and drug-related AEs were consistent with adverse reactions listed on the sapropterin package insert.31 An analysis of long-term safety in an extension study of multiple phase III studies of sapropterin 5–20 mg/kg/day found that most AEs were mild or moderate in severity and were unrelated to treatment.30 All growth parameters in the current study were slightly above the 50th percentile of the Centers for Disease Control and Prevention reference values at baseline and did not change significantly during the study ( Figure 3 ), indicating that sapropterin therapy for up to 2 years does not affect growth in young children with PKU.
Limitations
This 2-year interim analysis of an ongoing 7-year study was conducted using only the subset of patients for whom 2 years of follow-up data were available. As a single-arm, open-label study, there is no formal comparator arm, although there is relevant natural history in the literature. Furthermore, there are inherent challenges in analyzing scores from neurocognitive assessments repeated over time in an early childhood population. Patients began the study using one age-specific assessment and moved on to different age-specific assessments during the course of the study. The Bayley-III assesses early developmental performance in children age 0 to 30 months, and scores are not comparable with an IQ. Therefore, Bayley-III baseline data cannot be analyzed with later scores from WPPSI-III and WISC-IV.
Conclusions
Sapropterin reduced blood Phe levels and, in some cases, required an increase in prescribed dietary Phe intake. Sapropterin also had a favorable safety profile with no unexpected AEs in infants and young children with PKU. Neurocognitive function, as determined by mean change in FSIQ, was maintained over 2 years in children with PKU starting sapropterin therapy between 0 and 6 years of age, and the Bayley-III cognitive composite score indicated that no infant or toddler experienced developmental delay. Standard z-scores for height, weight, and head circumference were also maintained. Continued follow-up in this ongoing 7-year study will allow for evaluation of the long-term effects of sapropterin in children who began treatment at a very young age.
Appendix
Other investigators and sites participating in the PKU-015 study: M. Potter, McMaster University Medical Centre; B. Maranda, Centre Hospitalier Universitaire de Quebec—Genetics; K. McBride, Nationwide Children’s Hospital; L. Smith, Children’s Mercy Hospital; L. Casey, University of Alberta; P. Gordon, Penn State University; H. Levy, Children’s Hospital of Boston; W. Nyhan, University of California, San Diego School of Medicine; J. Phillips III, Vanderbilt University Medical Center; R. Chang, Children’s Hospital of Orange County; C. Greenberg, Children’s Hospital of Manitoba; S. McCandless, University Hospital Cleveland, Case Medical Center; J. Mitchell, McGill University Health Centre; A. Sanchez-Valle, Tampa General Hospital.
Disclosure
N.L., K.S., A.F., D.D., B.K.B., S.S., and S.W. are paid consultants of BioMarin Pharmaceutical. W.L., C.Z., and E.J. are employees of BioMarin Pharmaceutical and own stock and stock options in BioMarin. S.P. was an employee of BioMarin Pharmaceutical during the time of writing the manuscript and owned stock and stock options in BioMarin.
References
Scriver C, Kaufman S, Eisensmith RC, Woo SL . The hyperphenylalaninemias. In: Scriver CR, Sly WS, Valle D (eds.). The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill: New York, 1995:1015–1075.
Jervis GA . Phenylpyruvic oligophrenia (phenylketonuria). Res Publ Assoc Res Nerv Ment Dis 1954;33:259–282.
Paine RS . The variability in manifestations of untreated patients with phenylketonuria (phenylpyruvic aciduria). Pediatrics 1957;20:290–302.
National Institutes of Health Consensus Development Panel. National Institutes of Health Consensus Development Conference Statement: phenylketonuria: screening and management, October 16–18, 2000. Pediatrics 2001;108:972–982.
Holtzman NA, Kronmal RA, van Doorninck W, Azen C, Koch R . Effect of age at loss of dietary control on intellectual performance and behavior of children with phenylketonuria. N Engl J Med 1986;314:593–598.
Michals K, Azen C, Acosta P, Koch R, Matalon R . Blood phenylalanine levels and intelligence of 10-year-old children with PKU in the National Collaborative Study. J Am Diet Assoc 1988;88:1226–1229.
Mitchell JJ, Trakadis YJ, Scriver CR . Phenylalanine hydroxylase deficiency. Genet Med 2011;13:697–707.
Aldámiz-Echevarría L, Bueno MA, Couce ML, et al. Tetrahydrobiopterin therapy vs phenylalanine-restricted diet: impact on growth in PKU. Mol Genet Metab 2013;109:331–338.
Cunningham A, Bausell H, Brown M, et al. Recommendations for the use of sapropterin in phenylketonuria. Mol Genet Metab 2012;106:269–276.
Rohr FJ, Munier AW, Levy HL . Acceptability of a new modular protein substitute for the dietary treatment of phenylketonuria. J Inherit Metab Dis 2001;24:623–630.
Moyle JJ, Fox AM, Bynevelt M, Arthur M, Burnett JR . A neuropsychological profile of off-diet adults with phenylketonuria. J Clin Exp Neuropsychol 2007;29:436–441.
Stemerdink BA, Kalverboer AF, van der Meere JJ, et al. Behaviour and school achievement in patients with early and continuously treated phenylketonuria. J Inherit Metab Dis 2000;23:548–562.
Viau KS, Wengreen HJ, Ernst SL, Cantor NL, Furtado LV, Longo N . Correlation of age-specific phenylalanine levels with intellectual outcome in patients with phenylketonuria. J Inherit Metab Dis 2011;34:963–971.
Gassió R, Fusté E, López-Sala A, Artuch R, Vilaseca MA, Campistol J . School performance in early and continuously treated phenylketonuria. Pediatr Neurol 2005;33:267–271.
Moyle JJ, Fox AM, Arthur M, Bynevelt M, Burnett JR . Meta-analysis of neuropsychological symptoms of adolescents and adults with PKU. Neuropsychol Rev 2007;17:91–101.
Brumm VL, Azen C, Moats RA, et al. Neuropsychological outcome of subjects participating in the PKU adult collaborative study: a preliminary review. J Inherit Metab Dis 2004;27:549–566.
Chang PN, Gray RM, O’Brien LL . Patterns of academic achievement among patients treated early with phenylketonuria. Eur J Pediatr 2000;159(suppl 2):S96–S99.
Feillet F, MacDonald A, Hartung Perron D, Burton B . Outcomes beyond phenylalanine: an international perspective. Mol Genet Metab 2010;99(suppl 1):S79–S85.
Gassió R, Artuch R, Vilaseca MA, et al. Cognitive functions in classic phenylketonuria and mild hyperphenylalaninaemia: experience in a paediatric population. Dev Med Child Neurol 2005;47:443–448.
Janzen D, Nguyen M . Beyond executive function: non-executive cognitive abilities in individuals with PKU. Mol Genet Metab 2010;99(suppl 1):S47–S51.
Waisbren SE, Noel K, Fahrbach K, et al. Phenylalanine blood levels and clinical outcomes in phenylketonuria: a systematic literature review and meta-analysis. Mol Genet Metab 2007;92:63–70.
Burton BK, Grange DK, Milanowski A, et al. The response of patients with phenylketonuria and elevated serum phenylalanine to treatment with oral sapropterin dihydrochloride (6R-tetrahydrobiopterin): a phase II, multicentre, open-label, screening study. J Inherit Metab Dis 2007;30:700–707.
Lee P, Treacy EP, Crombez E, et al. Safety and efficacy of 22 weeks of treatment with sapropterin dihydrochloride in patients with phenylketonuria. Am J Med Genet A 2008;146A:2851–2859.
Levy HL, Milanowski A, Chakrapani A, et al. Efficacy of sapropterin dihydrochloride (tetrahydrobiopterin, 6R-BH4) for reduction of phenylalanine concentration in patients with phenylketonuria: a phase III randomised placebo-controlled study. Lancet 2007;370:504–510.
Trefz FK, Burton BK, Longo N, et al. Efficacy of sapropterin dihydrochloride in increasing phenylalanine tolerance in children with phenylketonuria: a phase III, randomized, double-blind, placebo-controlled study. J Pediatr 2009;154:700–707.
Leuret O, Barth M, Kuster A, et al. Efficacy and safety of BH4 before the age of 4 years in patients with mild phenylketonuria. J Inherit Metab Dis 2012;35:975–981.
Keil S, Anjema K, van Spronsen FJ, et al. Long-term follow-up and outcome of phenylketonuria patients on sapropterin: a retrospective study. Pediatrics 2013;131:e1881–e1888.
Hennermann JB, Bührer C, Blau N, Vetter B, Mönch E . Long-term treatment with tetrahydrobiopterin increases phenylalanine tolerance in children with severe phenotype of phenylketonuria. Mol Genet Metab 2005;86(suppl 1):S86–S90.
Lambruschini N, Pérez-Dueñas B, Vilaseca MA, et al. Clinical and nutritional evaluation of phenylketonuric patients on tetrahydrobiopterin monotherapy. Mol Genet Metab 2005;86(suppl 1):S54–S60.
Burton BK, Nowacka M, Hennermann JB, et al. Safety of extended treatment with sapropterin dihydrochloride in patients with phenylketonuria: results of a phase 3b study. Mol Genet Metab 2011;103:315–322.
Kuvan [sapropterin dihydrochloride] Tablet Prescribing Information. BioMarin Pharmaceutical, Inc: Novato, CA, 2007.
Acknowledgements
We thank the PKU-015 study investigators, study coordinators, and study site support staff. We also thank Laurie LaRusso for her contribution to the writing of the manuscript with funding provided by BioMarin Pharmaceutical; Douglas Okamoto (consultant), for biostatistics; Linda Osburn-Welsh (consultant) for project management; and the following BioMarin Pharmaceutical employees: Alzata Evans-Jones, Robin McMahon, and Katie Workman (study administration and management), Sheri Oakes and Ivan Valverde (clinical data management), and Dan DiPrimeo and Felicia Fong (statistical programming). We especially thank all the children and families who took part in and are continuing to take part in this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This work is licensed under a Creative Commons Attribution 3.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/3.0/
About this article

Cite this article
Longo, N., Siriwardena, K., Feigenbaum, A. et al. Long-term developmental progression in infants and young children taking sapropterin for phenylketonuria: a two-year analysis of safety and efficacy. Genet Med 17, 365–373 (2015). https://doi.org/10.1038/gim.2014.109
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2014.109
Keywords
This article is cited by
-
Diagnostic and therapeutic recommendations for the treatment of hyperphenylalaninemia in patients 0–4 years of age
Orphanet Journal of Rare Diseases (2018)
-
Development of a synthetic live bacterial therapeutic for the human metabolic disease phenylketonuria
Nature Biotechnology (2018)
-
The complete European guidelines on phenylketonuria: diagnosis and treatment
Orphanet Journal of Rare Diseases (2017)
-
Efficacy, safety and population pharmacokinetics of sapropterin in PKU patients <4 years: results from the SPARK open-label, multicentre, randomized phase IIIb trial
Orphanet Journal of Rare Diseases (2017)
