
Abstract
Purpose:
The purpose of this study was to undertake thorough genetic analysis of the bone morphogenetic protein type 2 receptor (BMPR2) gene in patients with pulmonary arterial hypertension.
Methods:
We conducted a systematic analysis for larger gene rearrangements together with conventional mutation analysis in 152 pulmonary arterial hypertension patients including 43 patients diagnosed as having idiopathic pulmonary arterial hypertension and 10 diagnosed as having familial pulmonary arterial hypertension.
Results:
Analysis of the BMPR2 gene revealed each of the four kinds of nonsense and frameshift mutations, one missense mutation, one splice-site mutation, and two types of exonic deletion. For cases in which exons 1–3 were deleted, the 5′ and 3′ break points were located in the AluY repeat sequences in the 5′ side of the adjacent NOP58 gene and in the AluY repeat sequences in intron 3, suggesting an AluY-mediated nonallelic homologous recombination as the mechanism responsible for the deletion. For the case in which exon 10 was deleted, nonhomologous recombination took place between the AluSx site in intron 9 and a unique sequence in intron 10.
Conclusion:
Exonic deletions of BMPR2 account for at least part of BMPR2 mutations associated with heritable pulmonary arterial hypertension in Japan, as previously reported in other populations. One of our cases was mediated via Alu-mediated nonallelic homologous recombination and another was mediated via nonhomologous recombination.
Genet Med 15 12, 941–947.
Similar content being viewed by others


Main
Pulmonary arterial hypertension (PAH) is a serious disease with a poor prognosis. Possible etiological factors for PAH development include vasospasm, endothelin overactivity, intimal hyperplasia due to increased growth factor secretion, and thrombus formation in small pulmonary arteries.1 The presence of familial disease in ~6% of PAH patients with no obvious secondary causes of PAH suggested that genetic factors play an important role in a substantial proportion of patients with PAH. Linkage analysis and positional cloning have identified mutations in the bone morphogenetic protein type 2 receptor (BMPR2) gene in ~60% of familial PAH (FPAH) cases and 10–40% of patients with idiopathic PAH (IPAH).2,3,4 BMPR2 mutations were also reported in FPAH patients and in 40% of the IPAH cases in a Japanese population,5 corresponding to the clinical observation that PAH prevalence does not vary among different races.6
Recent studies have clarified the presence of genome-wide copy-number variations caused by genomic rearrangement such as deletion, duplication, inversion, and translocation of genetic codes spanning more than 1,000 base pairs, with genetic rearrangements of BMPR2 in patients with FPAH resulting in deletion or duplication of one or more exons of the gene.7,8 Another study identified exonic deletion or duplication in BMPR2 in both FPAH and IPAH cases.9 These findings have demonstrated that BMPR2 mutations may account for a higher proportion of PAH patients than previously expected.
Genome-wide analysis of copy-number variations has indicated significant variation in distribution and frequency among populations with different ethnic backgrounds, although such information about the genetic rearrangements of BMPR2 has been limited to the data obtained. Furthermore, deletion break points in exonic deletions of BMPR2 have not been reported. We therefore undertook a thorough genetic analysis of BMPR2 in Japanese patients with PAH and investigated the deletion break points in exonic deletions of BMPR2.
Materials and Methods
Study population
The study included PAH patients and their family members in Kyorin University Hospital, Tokyo, Japan, who were enrolled from August 2010 to June 2012. The enrolled group comprised 43 patients with IPAH, 10 with FPAH (5 families), 20 with collagen-associated PAH, 44 with chronic thromboembolic pulmonary hypertension, 7 with Eisenmenger syndrome, 2 with pulmonary veno-occlusive disease, 2 with cor pulmonale, and 24 with other etiologies. This study was approved by the ethics committee of Kyorin University School of Medicine, and written informed consent was obtained from all patients and family members.
In this study, the definition of heritable PAH (HPAH) is as follows: HPAH includes FPAH in which at least two patients in a family have been diagnosed on clinical criteria and also includes sporadic PAH cases who carry BMPR2 mutation.
DNA sequencing of BMPR2 exons
DNA was isolated from patient blood samples by a previously reported method,10 and DNA concentrations were measured using a Qubit fluorometer (Life Technologies, Carlsbad, CA). The protein-coding sequence of BMPR2 from exon 1 to 13 was amplified by PCR using primers derived from the intron sequence (NCBI reference sequence NC_000002.11) and designed by the online Primer Blast resource (the sequences are available on request to the corresponding author). PCR was carried out using AmpliTaq Gold (Life Technologies) as follows: 1 cycle at 95 °C for 10 min; 40 cycles at 96 °C for 1 min, 56 °C for 15 s, and 72 °C for 45 s; and 1 cycle at 72 °C for 7 min. After agarose electrophoresis (3%), amplified fragments were purified by a MultiScreen Filter Plate (EMD Millipore, Billerica, MA) and then sequenced directly to detect point mutations in BMPR2 using an Life Technologies 3130xl genetic analyzer and Big Dye Terminator Cycle Sequence Kit (ver. 3.1; Life Technologies). The protein-coding sequence of activin-like kinase receptor 1 and endoglin gene was analyzed as described above.
Detection of BMPR2 exonic deletion and break points
BMPR2 exonic deletion was detected by multiplex ligation-dependent probe amplification using a Salsa multiplex ligation-dependent probe amplification kit (no. P093; MRC-Holland, Amsterdam, The Netherlands) and a 3130xl genetic analyzer, as previously reported.8 Multiplex ligation-dependent probe amplification data were analyzed on the basis of several control data from the manufacturer-recommended Excel-based spreadsheet and finally sorted according to exon order.
Break points in exonic deletions were examined using a combination of semiquantitative PCR, long PCR, restriction fragment analysis, and direct sequencing. Semiquantitative PCR was carried out using KOD plus polymerase (TOYOBO, Japan) under the following conditions: 1 cycle at 94 °C for 2 min and 26 cycles at 94 °C for 15 s, 56 °C for 30 s, and 68 °C for 45 s. Long PCR was carried out using PrimeSTAR GXL polymerase (TAKARA BIO, Otsu, Japan) under the following conditions: 30 cycles at 98 °C for 10 s and 66 °C for 10 min. PCR products and their restriction fragments (AatII, BamHI, HindIII, and PpuMI) were analyzed by agarose gel electrophoresis (0.7%), followed by another cycle of PCR analysis to identify the deletion break point. The sequence reaction was carried out as described above.
Results
Point mutations in BMPR2
Among IPAH and FPAH patients and their respective family members, each of the four kinds of nonsense and frameshift mutations, one missense mutation, one splice-site mutation, and two types of exonic deletion on BMPR2 gene, were found. Finally, 20 individuals (12 families) including 3 individuals who do not have obvious symptoms of PAH were identified as having HPAH ( Table 1 ). Only two kinds of point mutations were previously reported.11 However, point mutations in BMPR2 were not recognized in all of the cases with other etiologies, such as collagen-associated PAH, chronic thromboembolic pulmonary hypertension, Eisenmenger syndrome, and pulmonary veno-occlusive disease. No mutation was found in the activin-like kinase receptor 1 or endoglin genes (data not shown).
Exonic deletions in BMPR2
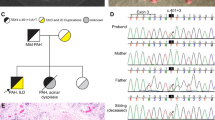
Figure 1 shows the multiplex ligation-dependent probe amplification analysis for exonic deletions of BMPR2. Two patients with FPAH (patients 11 and 11-2 in Table 1 ) showed deletion of exons 1–3, suggesting no transcript from this allele. Patient 11-3 was also diagnosed as having PAH at 17 years of age and died in another hospital. Two sibs of patient 11-2 did not have a deleted allele (data not shown).

Multiplex ligation-dependent probe amplification (MLPA) analysis of patients 11 and 12. (a) Family chart of patient 11 (Nos. 11 and 11-2 in Table 1 ). The sister of patient 11 (No. 11-3) had been diagnosed as having pulmonary arterial hypertension, and she had already died. (b) Raw data of MLPA analysis on the bone morphogenetic protein type 2 receptor (BMPR2) gene of patient 11. Exonic deletions of exons 1–3 in BMPR2 are shown by arrows. (c) The processed data of the MLPA analysis on the BMPR2 gene of patient 11. The MLPA analysis shows an exon 1–3 deletion (arrow). (d) Family chart of patient 12. (e) The processed data of MLPA on the BMPR2 gene of patient 12. The MLPA analysis shows the exon 10 deletion (arrow).
One patient (patient 12 in Table 1 ) showed deletion of exon 10 ( Figure 1e ), and the mother of patient 12 did not have a deleted allele, suggesting that the deleted allele was transmitted from the paternal side or by de novo deletion. Exon 10 deletion causes loss of the C-terminal portion of the kinase domain and cytoplasmic tail from the BMPR2 protein. Meanwhile, exonic deletions were not recognized in all of the cases with other etiologies.
Detection of deletion break points
In cases of exon 1–3 deletion (patients 11 and 11-2), semiquantitative PCR analysis revealed the lack of one copy of the 5′ adjacent NOP58 gene but the presence of two copies of SUMO1, a gene located further upstream. Therefore, 5′ break points were predicted between NOP58 and SUMO1. Furthermore, several forward primers in the intergenic region between NOP58 and SUMO1, and several reverse primers within the 45-kb BMPR2 intron 3 were designed, and PCRs were carried out. This strategy enabled bridging of the deletion, achieving a 2.2-kb fragment ( Figure 2a ). Sequence analysis on this fragment revealed a 225-kb deletion and the 5′ and 3′ break points, both mapping within the AluY repetitive sequence.

Deletion break point of patients 11 and 12. (a) Deletion break point of patient 11. Top: genomic structure of BMPR2 gene and 5′ adjacent region. Middle: genomic map of break points. Triangles and boxes with numbers indicate Alu sequences and exons. Arrows indicate PCR primers to bridge the deletion. Bottom: sequence profile of break and rejoining points. Inset: gel electrophoresis of bridge clone. (b) Deletion break point of patient 12. Top: genomic structure of BMPR2 gene exon 10 and neighboring region. Middle: genomic map of break points. Triangles indicate Alu sequences. Arrows indicate PCR primers used to bridge the deletion. Bottom: sequence profile of break and rejoining points. Inset: gel electrophoresis of bridge clone.
In the case of exon 10 deletion (patient 12), break points were located in intron 9 (9 kb) and intron 10 (11 kb). Long PCR between exon 9 and exon 11 obtained a 15-kb fragment, suggesting a 5-kb deletion including exon 10 ( Figure 2b ). The restriction analysis, nested PCR, and primer walking on this fragment revealed 5′–break point maps on AluSx in intron 9 and 3′–break point maps on a unique sequence within the Alu cluster in intron 10.
Discussion
This study demonstrated that (i) BMPR2 point mutations and exonic deletions may account for at least part of BMPR2 mutations associated with PAH in a Japanese population and (ii) exonic deletions associated with HPAH arise through the mechanism of not only Alu-mediated nonallelic homologous recombination (NAHR) but also nonhomologous recombination.
One type of mutation in exon 3 recognized in HPAH sisters (patients 4 and 4-2) and one splice-site mutation (patient 7) in this study were the same as reported in Caucasian cases,11 whereas other mutations detected in HPAH were novel. The exonic deletion of exon 10 was also reported previously,9 although it is not clear whether their deletions are identical to those in our results, because break points were not reported in the previous study.
One patient in this study (patient 8) showed a missense mutation in exon 8 (c.1016T>A/p.Val339Asp). Her daughter (patient 8-2) had some obvious symptoms of PAH since 14 years of age and, thus, had been diagnosed as having pediatric PAH. Our preliminary investigation revealed that the frequency of c.1016T>A is <1 of 200 chromosomes in the Japanese general population (data not shown). Val339 in BMPR2 is located in the “catalytic domain of protein kinase” and conserved across many vertebrate species. It is also conserved across the human transforming growth factor-β receptor family, although some family members such as BMPR1A and BMPR1B show replacement with the synonymous amino acid isoleucine. Such mutation of an uncharged amino acid to a negatively charged one in exon 8 is likely to impair the protein kinase activity of BMPR2, although we cannot rule out the possibility that an additional abnormality may account for this PAH mutation.
This study is the first to identify the exact break points of PAH exonic deletion. In one case, we demonstrated AluY-mediated NAHR and a large deletion (>200 kb) involving BMPR2-5′ exons and the 5′-neighboring gene NOP58 ( Figure 3a ). Therefore, the AluY-mediated deleted/duplicated allele of NOP58 and exons 1–3 can be expected ( Figure 4a ). It seems less possible that NOP58, a ribonucleoprotein homologue to yeast, is involved in the etiology of PAH because although NOP58 is widely distributed in many animal species, there is no positive evidence of irreplaceable function. NAHR-mediated deletions, duplications, inversions, and other alterations, especially Alu-mediated NAHR, have been implicated not only in copy-number variations within the human genome but also in numerous human genetic disorders.12,13 Therefore, the same type of NAHR could be responsible for other exonic deletions in PAH.

Dot matrix plot of break points. (a) Break point of patient 11. (b) Break point of patient 12. Triangles in x and y axes indicate Alu sequences in break point–neighboring regions. In cases in which DNA sequences in x and y axes are the same, dots are inserted in this plot field. Therefore, when repeated sequences such as Alu exist in x and y axes, lines appear because dots connect. The direction of the lines indicate direct repeat or invert repeat. Break points are located in the middle of the x and y axes. In patient 11 (a), the recombination point exists only on the dot line indicating direct repeat of Alu sequences, suggesting homologous recombination between Alu sequences. In patient 12 (b), the recombination point exists on the Alu sequence (AluSx) in intron 9, but it exists in between Alu sequences including some retrograded ones in intron 10, suggesting nonhomologous recombination.

Potential mechanisms of Alu -mediated exonic deletions. (a) Possible Alu-mediated nonallelic homologous recombination in family 11. In patients 11 and 11-2, AluY-mediated deleted/duplicated allele of NOP58 and exons 1–3 can be expected. (b) Possible Alu-mediated nonhomologous recombination in patient 12. Nonhomologous recombination between Alu sequences can induce a lost fragment and deleted/duplicated allele of exon 10. (c) Dot matrix plot of areas neighboring break points in patient 12. This plot shows a wider range in x axis than that in Figure 3 , indicating interesting homologous regions in both 5′ and 3′ sites in intron 10. (d,e) Sequence alignment of two possible Alu pairings close to a break point line in patient 12. (f) Sequence alignment of possible high but nonhomologous AluSx3/AluSx1 in 3′ sites in patient 12. (g) Alu sequences in areas of exon 10 (upper) and intron 10 (lower) in patient 12. Dot plot and sequence alignment analyses of these regions allow us to raise two possible models to produce a deleted allele. (h) AluJo/AluSx recombination and erroneous rejoining model in patient 12. One possibility is that AluSx1/AluSx3 nonhomologous pairing (f) triggers hairpin loop formation and AluJo/AluSx pairing (d). Then, accidental breakage in the middle of the hairpin, AluJo/AluSx recombination, and erroneous rejoining can produce a deleted allele. (i) AluSx3/AluSx recombination and erroneous rejoining model in patient 12. Another possibility is that AluSx1/AluSx3 nonhomologous pairing (f) triggers hairpin loop formation and AluSx3/AluSx nonhomologous recombination (e).
The second case of exon 10 deletion in this study, however, was not due to NAHR but instead to nonhomologous recombination between Alu-rich regions ( Figure 3b ). Alu-mediated NAHR has been attracting attention recently and was reported in association with selected diseases.14 Alu sequences are apparently involved in this recombination because the 5′-break point is in the middle of the AluSx-repetitive sequence. However, the adjacent sequence of 3′ break point in intron 10 seems to be unique, i.e., no homology to the 5′ break point or the adjacent region in intron 9 ( Figure 3b ). It is therefore possible that a two-step recombination is involved, first Alu–Alu NAHR and then possibly accidental nonhomologous recombination. Although there are many alternative models, the deleted allele is accompanied by a duplicated allele and a lost fragment. The possible ways of producing those models are simultaneous Alu–Alu pairings, hairpin loop formation and accidental break, and erroneous rejoining ( Figure 4b – i ). Our results raise the possibility that Alu-mediated, but not NAHR-mediated, accidental nonhomologous recombination occurs in some diseases at a certain level of frequency. Further analyses of other cases are needed to clarify the mechanism underlying this type of nonhomologous recombination.
We performed a preliminary semiquantitative reverse-transcriptase PCR to investigate the expression levels of BMPR2 mRNA (Supplementary Figure S1 online). As expected, the expression levels of BMPR2 mRNA were much decreased in patients 11 and 11-2 as compared with their family members, 11-3 and 11-4, without a deletion. Of note, there was no evidence of aberrant mRNA, and the level of intact BMPR2 mRNA was decreased in patient 12. This suggests that the transcript from the exon 10–deleted allele is depleted or instable, and it can be expected that no aberrant BMPR2 protein is translated. Therefore, both of these exonic deletions seem to cause stoichiometric imbalance due to haploinsufficiency, indicating low probability of a dominant-negative effect on BMPR2 function. However, these findings are based on preliminary data, and the quantification of mRNA by additional experiments such as real-time PCR is desired in the future.
Alu is a retrotransposon and the most repeated sequence in the human genome (about 10% of all the genome). The following mechanisms regarding Alu-mediated genetic disorders have been demonstrated: (i) deleted and duplicated allele is induced by homologous recombination between different Alu sequences mainly in introns;15,16 (ii) inactivation of genes is induced by insertion of Alu sequences into genes, in particular, into exons;17and (iii) variant Alu sequences in introns induce exonization.18 In particular, because Alu sequences share high homology with each other and their length is ~300 bases although they have grown in diversity along the course of evolution, the different Alu sequences are subject to homologous recombination. These findings suggest that Alu has an important role in genomic diversity. In this study, we demonstrated that PAH is one of the Alu-mediated genetic disorders and that the mechanisms could be mediated through both Alu-mediated NAHR and nonhomologous recombination. In the near future, the further detailed studies investigating the relationship between loss of BMPR2 function mediated by Alu and BMP signaling would be desirable using specimens from lung tissue of PAH patients.
In conclusion, exonic deletions of BMPR2 found in HPAH were mediated through both Alu-mediated NAHR and nonhomologous recombination.
Disclosure
The authors declare no conflict of interest.
Change history
03 February 2015
Masaharu Kataoka, Yuki Aimi, Ryoji Yanagisawa, Masae Ono, Akira Oka, Keiichi Fukuda, Hideaki Yoshino, Toru Satoh & Shinobu Gamou Genet Med 2013; doi:10.1038/gim.2013.41 Published online 11 April 2013 In the published version of this article, the position of a green bar in Figure 2a next to the box labeled NOP58 is wrong.
References
Rich S, Dantzker DR, Ayres SM, et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med 1987;107:216–223.
Morse JH, Jones AC, Barst RJ, Hodge SE, Wilhelmsen KC, Nygaard TG . Mapping of familial primary pulmonary hypertension locus (PPH1) to chromosome 2q31-q32. Circulation 1997;95:2603–2606.
Deng Z, Morse JH, Slager SL, et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet 2000;67:737–744.
Elliott CG . Genetics of pulmonary arterial hypertension: current and future implications. Semin Respir Crit Care Med 2005;26:365–371.
Morisaki H, Nakanishi N, Kyotani S, Takashima A, Tomoike H, Morisaki T . BMPR2 mutations found in Japanese patients with familial and sporadic primary pulmonary hypertension. Hum Mutat 2004;23:632.
Rich S . Heart Disease. WB Saunders Company: Philadelphia, 2001:1908–1935.
Cogan JD, Vnencak-Jones CL, Phillips JA 3rd, et al. Gross BMPR2 gene rearrangements constitute a new cause for primary pulmonary hypertension. Genet Med 2005;7:169–174.
Cogan JD, Pauciulo MW, Batchman AP, et al. High frequency of BMPR2 exonic deletions/duplications in familial pulmonary arterial hypertension. Am J Respir Crit Care Med 2006;174:590–598.
Aldred MA, Vijayakrishnan J, James V, et al. BMPR2 gene rearrangements account for a significant proportion of mutations in familial and idiopathic pulmonary arterial hypertension. Hum Mutat 2006;27:212–213.
Thomson JR, Machado RD, Pauciulo MW, et al. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-beta family. J Med Genet 2000;37:741–745.
Machado RD, Aldred MA, James V, et al. Mutations of the TGF-beta type II receptor BMPR2 in pulmonary arterial hypertension. Hum Mutat 2006;27:121–132.
Sasaki M, Lange J, Keeney S . Genome destabilization by homologous recombination in the germ line. Nat Rev Mol Cell Biol 2010;11:182–195.
Konkel MK, Batzer MA . A mobile threat to genome stability: the impact of non-LTR retrotransposons upon the human genome. Semin Cancer Biol 2010;20:211–221.
Zhang F, Seeman P, Liu P, et al. Mechanisms for nonrecurrent genomic rearrangements associated with CMT1A or HNPP: rare CNVs as a cause for missing heritability. Am J Hum Genet 2010;86:892–903.
Zhang G, Fukao T, Sakurai S, Yamada K, Michael Gibson K, Kondo N . Identification of Alu-mediated, large deletion-spanning exons 2-4 in a patient with mitochondrial acetoacetyl-CoA thiolase deficiency. Mol Genet Metab 2006;89:222–226.
Fukao T, Zhang G, Rolland MO, et al. Identification of an Alu-mediated tandem duplication of exons 8 and 9 in a patient with mitochondrial acetoacetyl-CoA thiolase (T2) deficiency. Mol Genet Metab 2007;92:375–378.
Apoil PA, Kuhlein E, Robert A, Rubie H, Blancher A . HIGM syndrome caused by insertion of an AluYb8 element in exon 1 of the CD40LG gene. Immunogenetics 2007;59:17–23.
Purevsuren J, Fukao T, Hasegawa Y, Fukuda S, Kobayashi H, Yamaguchi S . Study of deep intronic sequence exonization in a Japanese neonate with a mitochondrial trifunctional protein deficiency. Mol Genet Metab 2008;95:46–51.
Acknowledgements
This work was supported in part by the Health and Labour Sciences Research Grant from the Ministry of Health, Labour, and Welfare of Japan; Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology; a grant from the medical department collaborative project of Kyorin University; and a fellowship from Kyorin University School of Health Sciences.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Figure S1
(PDF 25 kb)
Rights and permissions
About this article
Cite this article
Kataoka, M., Aimi, Y., Yanagisawa, R. et al. Alu-mediated nonallelic homologous and nonhomologous recombination in the BMPR2 gene in heritable pulmonary arterial hypertension. Genet Med 15, 941–947 (2013). https://doi.org/10.1038/gim.2013.41
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2013.41
Keywords
This article is cited by
-
Investigation of the mechanism of copy number variations involving the α-globin gene cluster on chromosome 16: two case reports and literature review
Molecular Genetics and Genomics (2023)
-
Dual loss of human POLQ and LIG4 abolishes random integration
Nature Communications (2017)
