
Abstract
Biobanks and archived data sets collecting samples and data have become crucial engines of genetic and genomic research. Unresolved, however, is what responsibilities biobanks should shoulder to manage incidental findings and individual research results of potential health, reproductive, or personal importance to individual contributors (using “biobank” here to refer both to collections of samples and collections of data). This article reports recommendations from a 2-year project funded by the National Institutes of Health. We analyze the responsibilities involved in managing the return of incidental findings and individual research results in a biobank research system (primary research or collection sites, the biobank itself, and secondary research sites). We suggest that biobanks shoulder significant responsibility for seeing that the biobank research system addresses the return question explicitly. When reidentification of individual contributors is possible, the biobank should work to enable the biobank research system to discharge four core responsibilities to (1) clarify the criteria for evaluating findings and the roster of returnable findings, (2) analyze a particular finding in relation to this, (3) reidentify the individual contributor, and (4) recontact the contributor to offer the finding. We suggest that findings that are analytically valid, reveal an established and substantial risk of a serious health condition, and are clinically actionable should generally be offered to consenting contributors. This article specifies 10 concrete recommendations, addressing new biobanks as well as those already in existence.
Genet Med 2012:14(4):361–384
Similar content being viewed by others


Introduction
An ongoing debate focuses on the question of whether researchers bear duties to analyze and offer back to research participants incidental findings (IFs) and individual research results (IRRs) generated in genetic and genomic research. Much less has been written on the responsibilities of biobanks collecting data and/or samples for use in genetic and genomic research over time. Yet biobanks—a term we use broadly here to cover a range of structured collections, including biorepositories and databases (see Appendix)—are now crucial engines of large-scale genetic and genomic research. They are the central part of what we will call a “biobank research system,” comprising primary research or collection sites, the biobank, and secondary research sites accessing biobank data or samples for further research. IFs and IRRs can arise at multiple points in this system. How they should be handled and the role of biobanks in facilitating this process are pressing concerns and the focus of this article.
Many of us participated in an earlier project (#1-R01-HG003178) that offered analysis and recommendations addressing the responsibilities of researchers and Institutional Review Boards (IRBs) when the researcher collecting data and samples discovers IFs of potential health or reproductive importance to the research participant. That project resulted in consensus recommendations1 published as part of a journal symposium issue including a large number of individual papers offering additional analyses. This follow-on project (#2-R01-HG003178) broadens the focus in two respects: we consider management of IRRs as well as IFs, and we concentrate on the problem of how to identify and manage them in the increasingly important context of large-scale research involving biobanks.
Much genetic and genomic research now involves aggregating data and samples from multiple research projects or from clinical sources into a central database and biorepository for future research use. In addition, there are a growing number of biobanks built by directly collecting data and samples from a population or subpopulation to assemble a large-scale research resource. Both types of biobanks are important to genetic and genomic research. The large size of many of these collections facilitates the analysis of genetic variants that are rare, have modest association with phenotypic traits, or reveal such an association only when combined with other genetic variants.
Although the simpler example on which we focused in our earlier project, that of a researcher discovering IFs and IRRs in what we will call “primary research,” raises difficult questions that are still under debate, the more complex example of IFs and IRRs discovered by a biobank or by a researcher obtaining and analyzing biobank data or samples (in what we will call “secondary research”) raises still further questions. Literature and guidance are sparse on the responsibilities of biobanks when IFs or IRRs of significance are identified, and on how to address IFs and IRRs consistently across research networks spanning multiple institutions. Core questions include: What information, if any, about IFs and IRRs should be offered back to individuals whose genetic data and samples are stored in biobanks and archived data sets? Who has the responsibility of offering genetic/genomic research results to those individuals? What policies should be in place governing the research practices of both existing biobanks (which may have been assembled without attention to return of IFs and IRRs) and new biobanks?
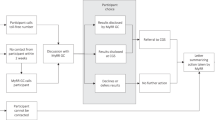
Figure 1 depicts primary researchers feeding data and samples into a biobank and secondary researchers accessing data/samples from the biobank for further research. This illustrates the relationship among these three types of entities: (i) primary researcher(s) or collection site(s), (ii) biobank, and (iii) secondary researcher(s). Together these entities comprise a biobank research system, which is more encompassing than the biobank itself. Although biobanks differ considerably in their structure, the three-stage schema depicted in Figure 1 is common and facilitates discussion of ethical responsibilities. Note that some biobanks collect the data and/or samples themselves, eliminating stage 1, the separate primary researchers/collection sites; this variation is depicted by the arrow running directly from contributors to the biobank. In addition, some biobanks perform research themselves without the involvement of secondary researchers, eliminating stage 3.

Depiction of primary researchers, biobank, and secondary researchers—a biobank research system. As noted in text, collection of data and samples from contributors may occur at Stage 1 research and collection sites or at Stage 2, the biobank itself.
Part I of this paper defines key concepts and specifies how IFs and IRRs arise in genetic and genomic research involving biobanks. Part II identifies the core questions to be addressed in deciding how to manage IFs and IRRs. We describe how biobanks appear to be addressing these issues now, and argue that biobanks have a responsibility to make their approach to IFs and IRRs explicit, whether or not the biobank plans to return the IFs and IRRs. We then address the design choice that biobanks face of irretrievably stripping identifiers versus preserving the possibility of reidentification and thus the return of individual IFs and IRRs to contributors. For biobanks in which reidentification—and thus return—is possible, we progress to considering what responsibilities biobanks should shoulder and offer recommendations for how those responsibilities should be discharged. We analyze the core responsibilities using the organizing acronym CARR (Clarifying, Analyzing, Reidentifying, and Recontacting): (1) clarifying the criteria for evaluating findings (e.g., analytic validity, seriousness of condition, and actionability) and the roster of returnable IFs and IRRs; (2) analyzing a particular finding in light of those criteria and that roster to determine whether it constitutes a returnable IF or IRR; (3) reidentifying the individual (or individuals) for potential return; and (4) recontacting the individual (or individuals) to offer the finding. Part III then crystallizes 10 specific recommendations. Part IV addresses the challenges posed by those recommendations for new biobanks versus biobanks already in existence.
This article advances the discussion of how to handle IFs and IRRs in genetic and genomic research by providing a systematic analysis of how IFs and IRRs should be handled in a biobank research system and clarifying how the CARR issues should be addressed at each stage of the process. We challenge the conventional view that would generally locate nearly all responsibilities with the primary researchers who collected the data and samples and place few, if any, responsibilities on the biobank itself. We suggest instead that biobanks should accept significant responsibilities for the management of IFs and IRRs. This article focuses on the return of IFs and IRRs in the context of research governed by US policies, regulations, and laws. We address these issues within the framework of current policy, regulation, and law, while acknowledging that the Department of Health and Human Services (DHHS) has invited comment on proposed changes to the Common Rule.2 Although we focus on the US context and rules, the debate over the return of IFs and IRRs is international. We refer to international sources where they illuminate key questions.
Methods
This project convened a multidisciplinary Investigator team and Working Group with expertise in genomic research and biobank management as well as medicine, law, bioethics, and empirical methods. The project conducted empirical research on current biobank policies to inform our normative recommendations. We refined a preliminary list of biobanks and searched the websites of all identified biobanks for policies on the return of IFs and IRRs. To supplement the search process, we also e-mailed the manager or director of identified biobanks and requested relevant documents. The results of that empirical research are reported by Johnson et al.3 We also conducted a comprehensive literature search, reviewing relevant regulations, guidance documents, and law. We developed a project website to share bibliographic materials and work products with the Working Group.
Over the course of the 2-year project, we held four Working Group meetings. In addition to Working Group members, the first meeting included a number of other National Institutes of Health (NIH)–funded researchers working on various aspects of return of IFs and IRRs, to compare approaches. The second meeting was held in Washington, DC, with participation of key researchers and personnel from NIH and the Centers for Medicare and Medicaid Services (CMS), which administers the Clinical Laboratory Improvement Amendments (CLIA). The third meeting included invited presentations on collected case studies, biobank structure and capacity to return IFs and IRRs, and international comparisons.
Before the fourth meeting, the principal investigator and two members of the project Working Group participated in a session on the return of IFs and IRRs at the annual meeting of the International Society for Biological and Environmental Repositories (ISBER), to elicit input from attendees. Our project then held a day-long public conference in Bethesda, Maryland, presenting our project’s draft recommendations and individual papers to elicit further feedback. The day after the conference, the Working Group met a fourth time to further refine our recommendations in light of the feedback received. Final revisions to these recommendations were negotiated by e-mail. As is common in consensus articles produced by large multidisciplinary author groups, individual authors may not agree with every point made below. However, we reached consensus on the thrust of this article and the article’s recommendations.
Part I: Definitions
A biobank is a structured resource that holds human biological samples and/or data to facilitate research over time. We use the term “biobank” broadly to cover a range of research resources that are sometimes referred to using different terms (see Appendix). We thus define biobank to include archived data sets as well as repositories of biological samples maintained for future research use. We also include biobanks set up to associate genotype information with phenotype, such as biobanks that link human genomics to electronic medical records, biobanks that link genomic data to neuroimaging scans or other nongenomic data sources, and biobanks that analyze environmental samples or data in relation to human genomics.
Biobanks have become crucial to the conduct of genetic and genomic research, especially large-scale genomic research. Biobanks now exist all over the world. Many have been established in the United States, with more planned. Biobanks vary considerably in size, scope, design, and intended use.4 A growing literature documents the variation, including large population-based biobanks (such as UK Biobank), biobanks located at a center of clinical care (such as biobanks in the Electronic Medical Records and Genomics (eMERGE) Network), and disease-specific biobanks (such as Myeloma Bank on a Cure).5,6,7,8,9,10 Biobanks vary in management and governance structure, so that precisely who discharges a responsibility falling on the biobank (including those responsibilities we recommend below) will vary; biobanks themselves are generally best situated to identify the relevant individual(s) within the biobank.
As noted above, some biobanks collect their own data and samples, while others rely on primary researchers and collectors at multiple sites to perform collection, with data and/or samples then aggregated in the biobanks. Three variables of importance to our analysis are: whether the biobank is itself conducting research, the degree and type of deidentification of data and samples in the biobanks, and (related to this) whether biobank research and secondary research by researchers accessing biobank data/samples qualify as research on human subjects under the Common Rule.11 We address these three variables in turn.
Some biobanks themselves conduct research; examples include disease-focused biobanks such as Myeloma Bank on a Cure and ongoing cohort studies such as the Framingham Heart Study.12 However, some biobanks merely aggregate data and samples from primary researchers or collection sites and supply those data and samples to secondary researchers, with no research conducted by personnel at the biobank itself. Both types of biobanks function as a central part of a larger biobank research system.
When biobanks receive deidentified data and samples that were collected for purposes other than the biobank’s research, the biobank may not be conducting human subjects research under the Common Rule.13,14,15 Whether the Common Rule applies affects the responsibilities of the biobank, including responsibilities for managing the IFs and IRRs (see Part II’s section on, “If data and samples can be reidentified, are there any biobank research system responsibilities to offer return of IFs and IRRs?”). The Common Rule specifies that research on human subjects is research using information that is “individually identifiable (i.e., the identity of the subject is or may be readily ascertained by the investigator or associated with the information)” (§46.102(f)).11 Excluded is research on data and samples when “subjects cannot be identified, directly or through identifiers linked to the subjects” (§46.101(b)(4)).11
In keeping with the Common Rule, the Office for Human Research Protections (OHRP) has advised that when researchers use data or samples that were not collected for that research and the source individuals cannot readily be identified by the researchers, this does not meet the regulatory definition of research involving “human subjects” (a term defined by the Common Rule at 45 C.F.R. § 46.102(f) to be a “living individual about whom an investigator…obtains data through intervention or interaction with the individual or identifiable private information”).13 Similarly, OHRP has stated that institutions are not engaged in human subjects research when they receive coded data or samples from another institution engaged in research that retains the code, and the receiving institution cannot readily reidentify the subjects.14 The predecessor of the OHRP, the Office for Protection from Research Risks, specifically addressed research using stored data or tissue, stating that “human subjects” are not involved when the material “was collected for purposes other than submission to the Repository,” and the “material is submitted to the Repository without any identifiable private data or information.”15 Thus, some biobank research constitutes human subjects research covered by the Common Rule, while other biobank research is what has been called “human non-subjects research” and is not covered by the Common Rule.16,17 Developing appropriate standards for the second type remains a challenge, one that DHHS attempts to address in a recent proposal of possible changes to the Common Rule.2
In part because some biobank research is considered human subjects research under the Common Rule while other research is not, a range of terms is used to refer to the human beings whose data and samples are housed in biobanks. Such an individual may be called a “participant” or “human subject” when the research being conducted qualifies as human subjects research, or the individual may be called a “donor” or “source” of the data or samples. Because we address both human subjects research and “human non-subjects research,” we use a more encompassing term than “subjects” or “participants.” We generally use the term “contributors” to refer to the individuals whose data and samples are collected in biobanks, whether or not they also qualify as “human subjects” under the Common Rule. We avoid the term “sources,” as this is ambiguous in a biobank research system in which the primary research or collection sites are institutional sources for biobank data and samples, and the biobank in turn is a source for secondary researchers. We also avoid the term “donors,” to avoid implying that the individuals whose data and/or samples are collected have donated a gift and are owed nothing in return (as some would argue in the case of whole-organ donation, for example). Although there is no perfect term, “contributor” suggests the vital contribution each individual makes to the assembly of a biobank and subsequent conduct of research.
Both IFs and IRRs can arise in primary research, biobank research, and secondary research. In our prior project, we defined an IF as “a finding concerning an individual research participant [or here, an individual contributor] that has potential health or reproductive importance and is discovered in the course of conducting research but is beyond the aims of the study.”1 In contrast, an IRR is a finding concerning an individual contributor that has potential health or reproductive importance and is discovered in the course of research, when the finding is on the focal variables under study in meeting the stated aims of the research project.
Some commentators have questioned the utility of distinguishing between IFs and IRRs, especially in the context of whole-exome, whole-genome, or genome-wide association studies (GWAS). We agree that the distinction between IFs and IRRs is the “fuzziest” in large-scale discovery research where it is difficult to identify what is “beyond the aims of the study” because the entire genome is under scrutiny and the research is inductive discovery research rather than research driven by discrete hypotheses.1,18,19 Moreover, recruitment for biobank participation may be for an open-ended array of future studies. However, IFs may still arise in ascertaining a prospective contributor’s eligibility to participate in the study or biobank and in baseline screening. For example, during initial enrollment, a researcher may discover that a contributor has elevated blood pressure—a finding incidental to the genomic study itself. IFs may also arise during the course of studies aiming to study a particular set of genotype/phenotype correlations. A researcher using a GWAS approach in a breast cancer study, for instance, may find mutations in genes known to be correlated with development of colon cancer.20 Because the colon cancer genetic association was beyond the aims of the breast cancer study, it is an IF.
Our research has uncovered biobanks, studies, and policies that indeed recognize a difference between IFs and IRRs. Yale University’s IRBs, for example, have generated policy recognizing that in some studies a determination may be made to return IFs but not IRRs.21 Policies contemplating return of IFs but not IRRs may be based on a number of factors, including anticipation that IRRs, as the focus of the research, may not be as well understood and validated, whereas many IFs may be fully validated and well-understood findings commonly communicated in clinical care. UK Biobank is an example of a biobank that offers contributors IFs (such as elevated blood pressure) discovered in baseline screening at enrollment, but not IRRs from the genetics/genomic analysis ensuing.22,23 In planning the cancer Human Biobank (caHUB), the National Cancer Institute (NCI) has contemplated the return of a finding of discrepant diagnosis when specimen pathology is checked upon submission to the biorepository and the diagnosis differs from that at the source site, but not necessarily the return of ensuing genetic and genomic research results.24 Indeed, any study or biobank collecting or archiving either medical information or environmental exposure information may stumble upon clinically significant information whose discovery was not the research aim, and thus may face the question of whether to return these IFs.
The distinction between IFs and IRRs thus serves a useful purpose in promoting the analysis of different types of findings. Table 1 systematizes how IFs and IRRs can arise in primary research or at the collection site, at the biobank, and in secondary research using biobank data and samples. This article maintains the distinction between IFs and IRRs, but recommends that biobanks, researchers, IRBs, and policy makers consider in each research context whether they can be distinguished and what the relationship between the return of IFs and IRRs should be, striving for a harmonized approach, especially when individual contributors may not readily perceive a distinction between these two types of findings. Differences in the handling of IFs and IRRs should be carefully explained.
Part II: Core Questions
How are biobanks handling IFs and IRRs now?
Although considerable debate has swirled around the issue of how to manage IFs and IRRs in human subjects research generally, there has been much less debate over biobank responsibilities. To ground our discussion of biobank responsibilities, we conducted research on current biobank policy. That research is described in greater detail by Johnson et al.3 In 2009–2010, we assembled a sample comprised of three types of biobanks: NIH intramural biobanks (43 biobanks), other US biobanks not coordinated intramurally through NIH (66 biobanks), and non-US biobanks (12 biobanks). A total of 2,366 documents were collected from the websites of the 43 NIH intramural and 12 non-US biobanks plus a random sample of 30 of the other US biobanks, for a total of 85 biobanks. Supplemental e-mail solicitations were sent to each biobank to request additional documents not publicly available on the biobank’s website. Forty-six biobanks responded to the request and sent a total of 29 additional documents, for a response rate of 54% to the e-mail request. Here, we briefly summarize relevant findings from the US biobanks only.
Of the US biobanks, almost half (43–49%, tabulated for the two categories of US biobanks) addressed issues related to the return of IFs and IRRs in some way. However, that means that more than half did not. Given how little literature exists offering guidance to biobanks on IFs and IRRs, it is not surprising that we found that many biobanks did not address the return of IFs and IRRs.
Of those biobanks addressing these issues, 33–46% had documents saying they do return some information. Among the 21 NIH intramural biobanks addressing whether they would return information to participants, seven of these had documents indicating that they would return some information (including GENEVA, the Framingham Heart Study, and the National Children’s Study). Among the 13 other US biobanks sampled and addressing this issue, six had documents indicating that they would return some information (including the Personalized Medicine Project at Marshfield Clinic, 23andMe, the University of Connecticut Behavioral Gene Bank, and the Kaiser Permanente Research Program on Genes, Environment, and Health). Table 2 , adapted from Johnson et al.,3 summarizes these findings.
Thus, we found that current biobank policies vary. The sample of US biobanks that responded to our inquiries was split fairly evenly on whether they even address the return of IFs and IRRs. Among those biobanks that do address this issue, some return no findings at all, some return nongenetic IFs (such as abnormal blood pressure) discovered at enrollment, some return a subset of nongenetic or genetic IFs, and some return a subset of nongenetic or genetic IRRs. Table 3 offers examples of biobanks with these different policies.
Do biobanks have responsibilities to address whether IFs and IRRs will be returned at all?
There is growing agreement that researchers have a responsibility to determine and make clear to research participants whether IFs and IRRs will or will not be offered back to the participants.20,25,26,27,28 As we noted in our prior project’s consensus article,1 in the face of silence, participants may simply assume that such findings will be returned. This assumption risks “conflat[ing] the role of researcher with that of a physician”29 and may lead participants to misconstrue researcher silence as an indication that there are no findings of individual health concern.
Similar problems may arise in biobank research systems. At least when contributors are asked to consent to use of their samples and/or data in a biobank research system (as opposed to those contributors never asked for consent because their samples and/or data are deidentified and used in research that is not considered human subjects research), the contributors may expect to be offered IFs and IRRs.29,30,31 This may be especially true when contributors consent to the use of their samples and/or data in biobank research focused on a particular disease. Biobank research systems need to make clear to contributors whether no IFs and IRRs will be returned or there is indeed an option for return. If there is an option for return, biobank research systems will need to ask whether individual contributors consent to recontact and return.
A further reason why biobank research systems should make clear their policy on return is that a growing body of empirical research suggests that many individuals want and even expect to receive their IFs and IRRs, especially if researchers find something serious.29,32,33,34,35,36 Thus, individuals may not only assume that they will be offered IFs and IRRs; they may desire such return. Individuals who want their results may feel their relationship with researchers should be reciprocal29,34,37 and that returning a serious finding should be a matter of “common courtesy.”34 Even though Beskow and Burke19 caution that the available studies do not reflect “participants’ nuanced preferences,” the evident desire for IFs and IRRs among many participants supports the importance of clarifying whether findings will be returned.
What design options are open to biobanks to handle IFs and IRRs? Designing to allow or to avoid reidentification
A range of design options is open to biobanks. The design question of whether to preserve the possibility of reidentification or make it impossible has direct implications for the ability to reidentify individual contributors and return IFs and IRRs. Thus, biobanks have to make a fundamental decision: (i) to design the biobank so that reidentification of contributors can be accomplished (at the primary research or collection site, at the biobank itself, or by a “trusted intermediary,” as discussed further below), thus enabling the return of IFs or IRRs, or (ii) to design the biobank so that reidentification is impossible because no one maintains the links to individual identities and thus IFs and IRRs cannot be returned. Only if reidentification is possible do biobanks face the further question of what return, if any, will be offered. A biobank that irretrievably strips identifiers and prohibits researchers from attempting to reidentify contributors (such as BioVU) cannot return IFs and IRRs.38,39 As Pulley et al.38 note, BioVU is “one of few biobanks” set up to conduct “nonhuman subjects research” and “the design explicitly precludes recontact with any individual.” Although reidentification may not be literally impossible40 and DHHS has recently noted that advancing technology may make de-identified data reidentifiable,2 BioVU is deliberately designed to block reidentification.
A decision to design a biobank by irretrievably stripping identifiers and retaining no link to identifiers, consequently precluding return, may be based on a number of factors. For example, Pulley et al.38 argue that BioVU’s design increases contributor privacy, thus “minimiz[ing] the risk of harm to individuals while maximizing the benefit to the broader society.”38 However, many (if not most) biobanks will be designed differently, for a range of research reasons. For example, the primary researchers or the biobank itself may follow contributors prospectively and indeed may seek to collect samples or data from contributors at multiple points in time. When biobanks receive samples and/or data from individual research sites that themselves maintain identifiers, then reidentification and return of IFs and IRRs can be accomplished. Moreover, there are biobanks that maintain the capacity themselves to reidentify contributors. Whenever contributors are reidentifiable within the biobank research system, the design is one in which IFs and IRRs can be returned. This leads to the challenging questions of what further choices these biobanks should make on the questions of whether to return IFs or IRRs and, if so, which ones and how.
If data and samples can be reidentified, are there any biobank research system responsibilities to offer return of IFs and IRRs?
The published literature reveals a growing number of commentators concluding that if return is possible, some findings should indeed be considered for return. Few commentators argue that there should be zero return, no matter how urgent and clinically actionable the finding. Our project members nonetheless discussed whether a biobank that could reidentify participants and thus offer results might decide as a blanket matter to offer no return at all. In the end, we decided to urge biobanks to address the IFs/IRRs issue and consider what findings would warrant an offer of return. There is considerable controversy over this last question—what findings merit an offer of return. Most of the literature to date, however, addresses return in individual studies or what we are calling “primary research,” not biobank research systems. We first briefly summarize the work on individual studies, the necessary starting point for considering the ethics of return in the more complex case of a biobank research system.
The ethics of return in individual studies. In our prior project, we analyzed ethical arguments on return of IFs in individual studies, concluding that researchers do indeed bear duties to manage IFs and offer a subset of them back to research participants consenting to return.1 We based that conclusion on several grounds. First, when the federal regulations stating standards for human subjects research apply (preeminently DHHS’s Common Rule,11 though the Food and Drug Administration also has regulations on human subjects 41,42), they impose requirements that are germane, even though the regulations do not explicitly address IFs and IRRs.11 For one, the regulations provide that consent must address research risks and potential benefits; IFs and IRRs may be seen as both risks and potential benefits, as they may impart crucial and even life-saving clinical information, but also may impose anxiety and the burdens of follow-up, with some IFs and IRRs turning out to be benign or even false positives.43 As IFs and IRRs do impose risks, the provisions calling for minimization of risk and that risks must be reasonable in relation to anticipated benefits are also germane.
Second, a number of ethical arguments support researcher responsibilities to manage and offer return of some IFs and IRRs. Richardson and Belsky,44 for example, argue that participant vulnerability and researcher fiduciary duties mean researchers owe a limited duty of “ancillary care” (care beyond that required to carry out the research safely), and Richardson45 has clarified that this encompasses a duty to offer back to the participant some IFs. They argue that when research participants entrust otherwise private information to researchers or provide researchers access to some aspect of the participant’s body, this “partial entrustment” carries with it certain researcher duties, including the duty to offer back information discovered of clinical importance. Miller et al.46 similarly argue that the researcher’s ethical obligation to return IFs is rooted in the researcher’s professional relationship with the participant, privileged access to private information about the participant, and discovery of an IF bearing on the participant’s health.
Illes et al.25 maintain that researcher duties to manage and offer the return of IFs flow from ethical duties to respect participant autonomy and interests. They suggest that researchers, whose work depends on the generosity of research participants and their willingness to be part of research, bear a duty of reciprocity. Kohane et al.47 argue that offering discoveries back to individual research participants allows them to be “partners in research rather than passive, disenfranchised purveyors of biomaterials and data.” The empirical literature to date shows that many individuals say they do indeed want to receive their IFs and IRRs, especially if researchers find something serious.29,32,33,34,35,36
In some respects, the international community has gone further than the United States in recognizing a duty to return IFs and IRRs in individual studies. Indeed, the IFs issue was acknowledged by the international community in the mid-1990s in a statement by the international Human Genome Organisation (HUGO), which declared that “choices to be informed or not with regard to results or incidental findings should…be respected.”48,49,50 In its “International Ethical Guidelines for Biomedical Research Involving Human Subjects,” the Council for International Organizations of Medical Sciences (CIOMS)51 has provided that “individual subjects will be informed of any finding that relates to their particular health status.” CIOMS also states that “subjects have the right of access to their data on demand, even if these data lack immediate clinical utility.”51
Some authors, however, have argued against return, or at least against return beyond the most urgent and actionable findings. These authors caution that research budgets are limited and should be devoted to the central aim of research, creating generalizable knowledge. They worry that return of individual findings will divert scarce resources, invite research participants to mistake research for clinical care, and may involve return of findings that are not yet adequately understood and validated.52 Yet even authors expressing concern over individual research findings tend to allow for return of some findings. Thus, within the United States and beyond, a growing number of authors maintain that researchers indeed should consider some subset of findings for return to research participants, even if there is not yet agreement on what set.1,19,53,54,55,56,57,58 Less discussed (especially in the United States) are the responsibilities of biobanks faced with these issues, as well as the responsibilities of what we are calling secondary researchers, though that conversation is beginning.19,24,57 We address the responsibilities of biobanks and secondary researchers in turn.
The ethics of return in biobank research systems. A robust and international ethics literature addresses ethical obligations of biobanks. Commentators query whether the complexities of biobank research call for expanding the roster of ethical precepts beyond those applicable to smaller-scale biomedical research.59,60 For example, some authors have explored the ethical obligations of genomic biobanks as public goods.61,62,63,64 Indeed, in some countries, biobanks are created by statute or structured as a nonprofit foundation or charitable organization.65 In the United States, biobanks created by NIH or another public entity (e.g., NIH’s dbGaP, or NCI’s caHUB) or funded publicly (e.g., the NIH-funded eMERGE Network) will bear public responsibilities accordingly. NCI, for example, in its Best Practices for biorepositories, stresses custodianship responsibilities: “responsible custodianship requires careful planning and transparent policies to ensure the long-term physical quality of the biospecimens, the privacy of human research participants, the confidentiality of associated data, and the appropriate use of biospecimens and data.”26 There is a significant literature on the public responsibilities of population biobanks.66,67,68,69
Privately funded biobanks (such as a biobank funded and created by a pharmaceutical company) bear some public responsibilities as well. ISBER’s Best Practices70 maintain that biobanks need to provide responsible “custodianship” of the tissues and data they collect, maintain, and share. And private biobanks owe duties to those individuals generous enough to participate by donating data or samples.71,72 Biobanks—private and public—commonly have a range of committees and governance structures to address operational and ethics issues including access to data and samples by secondary researchers.26,70,73
Core issues addressed in discussions of biobank ethics include consent and withdrawal of consent; protection of privacy and confidentiality; ownership of data and samples, benefit sharing, and commercialization; and sharing of data and samples with other researchers. Less discussed have been the ethical responsibilities of biobanks with respect to the return of IFs and IRRs. Boggio74 reports a division of view among respondents in a sample including US and international biobank experts: To “a substantial number of respondents…beneficence requires that information potentially of use to participants ought to be passed on to them….This trend is counteracted by the view—also popular among respondents—that returning research results is beyond a biobank’s duty and/or sharing results could do more harm than good.” The author notes, however, that several respondents suggested that preserving the capacity to return IRRs militated “in favor of forms of reversible anonymization of samples and associated data.”74
As noted above, a fundamental complexity attending biobank research is that some currently falls under the Common Rule and some does not. Biobanks that are collecting their own data and/or samples for research, as well as biobanks receiving data and/or samples collected for other purposes but conveyed with participant identifiers, will be conducting research on human subjects under the Common Rule. However, biobanks receiving data and/or samples that were not collected for the biobank and have identifiers removed, will not. A major caveat is that if samples or data are reidentified, the Common Rule will apply.16
When the Common Rule does apply, ethical duties will devolve on the biobank with implications for the return of IFs and IRRs. These will include the duty to address risks and potential benefits (if any) to those participating in the research; IFs and IRRs carry both. However, whether or not biobank research constitutes research involving human subjects under the Common Rule, there is increasing agreement that biobanks at least have the responsibility to address in their planning and in any informed consent documents whether IFs and IRRs will be offered back to individual contributors and, if so, how.26,65,75,76,77 For example, NCI’s 2011 Best Practices for Biospecimen Resources states, “The informed consent document should state whether individual or aggregate research results will be released to the human research participant, the participant’s healthcare provider, or the participant’s family and, if so, the mechanism for communicating such results….The procedure for opting out of all communications should be clearly indicated.”26
Secondary researchers accessing data and/or samples from the biobank will generally receive deidentified material that was not collected specifically for their research project and thus will be conducting research not covered by the Common Rule.13 This, combined with the fact that they will generally have no contact with contributors and may be far removed from data and sample collection in space and time, may lead some to conclude that secondary researchers have no responsibilities with respect to recognition of potentially returnable IFs or IRRs and their return. Yet this depicts secondary researchers in isolation rather than as part of the flow of data and samples through the biobank research system. Secondary researchers using biobank data or samples interact with the biobank. They apply for access to the data or samples, their application is reviewed and decided upon by the biobank’s Data Access Committee (DAC) or equivalent using explicit access criteria, negotiation may ensue over the terms of access, and access will be governed by an agreement, typically a Material Transfer Agreement (MTA) or Data Access Agreement (DAA; sometimes called a Data Use Agreement (DUA)). Thus, the biobank will have to determine prospectively the conditions of access and terms of these agreements. Terms already in use and relevant to the IFs/IRRs debate are that the secondary researcher will not seek to reidentify contributors and that research results will be communicated to the biobank.78,79,80,81
These terms suggest that when secondary researchers convey research results to the biobank, both the secondary researchers and the biobank will have an opportunity to spot those that should potentially be returned to contributors. Biobanks can anticipate this in their MTAs and DAAs or DUAs. The fact that secondary researchers are part of the larger biobank research system and that such researchers access and use data and samples according to biobank rules means that biobanks can address aspects of the IFs/IRRs problem in setting the terms of access and use. Specifically, biobanks can communicate any roster of returnable IFs/IRRs to secondary researchers; can ask those researchers applying for access to address whether the secondary research is likely to generate such IFs/IRRs and, if so, how they will be identified; and can require secondary researchers to communicate such IFs/IRRs back to the biobank so that they may be considered for return. Sharing with secondary researchers a concrete list of returnable findings may be the simplest and most efficient way to help secondary researchers flag any returnable IFs/IRRs.
Arguing that secondary research should simply lie beyond the reach of biobank responsibilities to handle IFs and IRRs ignores the fact that biobanks exist in large part to enable that secondary research. Much of what biobanks do is structure access to and use of the data and samples. Indeed, if a number of biobanks are already using MTAs and DAAs or DUAs to require secondary researchers to convey genomic results generated to the biobank, it is not a far stretch to ask that secondary researchers flag those results that are on a roster of potentially returnable IFs and IRRs. Ethical analysis of biobank-based research typically sees the biobank itself as the primary entity bearing duties of responsible custodianship.71,72,82 It thus makes sense that the biobank should set the rules for the overall process of recognizing (and subsequently analyzing and returning) IFs and IRRs, a process in which secondary researchers are asked to play a part.
Should the biobank itself have responsibilities for managing the return?
If IFs or IRRs are to be returned at all, the conventional view has been that the biobank has no responsibilities. Instead, primary investigators (or collection sites) should be responsible for identifying and contacting contributors, because primary researchers have the identification information and have had direct contact with contributors.24 According to NHGRI, if a secondary researcher “does generate results of immediate clinical significance,” that researcher “can only facilitate their return by contacting the contributing investigator who holds the key to the code that identifies the participant. In such cases, the contributing investigator would be expected to comply with all laws and regulations and consider the benefits and risks associated with the return of IRRs to participants and follow established institutional procedures (e.g., consultation with and approval by the IRB) to determine whether return of the results is appropriate and, if so, how it should be accomplished.”83
Placing responsibility for deciding whether and how to return IFs and IRRs on the original, collecting researcher, also comports with NIH policy on GWAS.84 Under that policy, “The NIH does not anticipate that participants will be able to obtain individual results of secondary analyses on data obtained from their participation in primary studies. Because the NIH GWAS data repository and secondary data users will not have access to identifying information or to the link to the keycode within the data, neither will be able to return individual results directly to subjects. Secondary investigators may share their findings with primary investigators, who may determine whether it is appropriate to return individual or aggregate research results to participants whose health may be affected....”84
Neither of these policies recognizes a role for the biobank itself. Instead, they place on primary researchers and their IRB the responsibility for determining whether to return IFs and IRRs at all, deciding which to return, and performing any actual return. In both cases, however, this approach is premised on the biobank and secondary researchers having no access to identifying information because primary researchers and institutions deidentified the data before submission to the biobank. It is important to recognize, though, that this is not the only model of biobank research. Biobanks can receive data and samples that are not deidentified, even if the biobank then removes identifiers before releasing the data and samples to secondary researchers. Indeed, a biobank—especially biobanks set up to study individuals with a particular disease or condition—may follow contributors prospectively, collecting additional information for research. When the biobank itself receives identifying information, the premise of these NHGRI and NIH policies does not apply. This reopens the question of what responsibilities the biobank should shoulder for deciding whether to return IFs and IRRs at all, determining which to return, and then managing the return process.
Moreover, even when deidentification by the primary researcher means that the biobank cannot reidentify and recontact the contributor, the question of biobank responsibilities remains. Reidentification and recontact are only the final phases of return of IFs and IRRs. Prior steps are deciding whether any IFs and IRRs should be returned, establishing the criteria to be used in ascertaining what findings are returnable, and analyzing particular findings to decide whether those specific IFs or IRRs should be returned. The fact that the biobank itself may not be able to reidentify and recontact does not answer the question of whether it bears some responsibility to determine whether any return will be offered, address the question of what criteria should be used to distinguish returnable findings, and analyze particular findings in the biobank’s research and possibly secondary research as well, as discussed below. In fact, a careful reading of both the NHGRI and NIH policies above shows that they recognize this potential division of labor. Both suggest that at least secondary investigators may indeed discover findings that raise the question of potential return, and both policies direct the secondary investigator to communicate the finding of concern to the primary investigator in order to consider return.
Thus, both policies suggest that the primary researcher may not bear sole responsibility for handling IFs and IRRs that emerge at the biobank and secondary research levels. Indeed, there are reasons to avoid placing all responsibilities on the primary researcher.85 The primary researcher may have limited knowledge and understanding of the biobank and secondary research and the meaning of the IFs/IRRs found there. Further, IFs or IRRs may be discovered by the biobank or secondary researchers years or decades after the original research has been completed. The primary researcher’s funding may be expired at that point. The primary researcher may actually be retired, departed from the institution, or deceased. Note that a recent consensus statement recommends that researcher obligations to manage IRRs end with the expiration of research funding, though the researcher may exercise his or her discretion to address IRRs beyond that point.86
At a July 2010 workshop convened by NCI, participants identified problems with the conventional view that primary responsibility to deal with IFs and IRRs should rest on the primary research or collection site. Because of these problems, some participants suggested that the biobank itself should have significant responsibility for addressing IFs and IRRs discovered at the biobank and in secondary research, rather than acting as a “passive entity.”24 Biobanks may also be able to marshal additional resources to cope with IFs and IRRs. Involving the biobank, rather than relying exclusively on individual collection and primary research sites, may additionally offer more consistency in how IFs and IRRs are handled for contributors whose data and samples are housed in a given biobank, no matter where they were collected.
Recognizing that biobanks bear responsibilities to address the IFs and IRRs question and should anticipate potential IFs/IRRs from biobank and secondary research would have several major implications. First, biobanks would need appropriate policy-making capacity to address the question of whether to return IFs and IRRs at all. If they decided to offer some return, they would need governance and oversight structures as well as policies to fulfill this function. They would also need to have the capacity to evaluate potential IFs and IRRs, managing the scientific and ethical issues arising. Biobanks conducting human subjects research will likely have an IRB already, but the challenge of the IFs and IRRs issues may suggest that other biobanks need an IRB (whether their own, or access to others’). The biobank’s IRB will need the capacity to interact with IRBs at primary research or collection sites, as offering findings back to an individual contributor may involve both IRBs. Biobanks may additionally want to create a specialized committee for management of return of IFs and IRRs (if any are offered), one model being the Informed Cohort Oversight Board (ICOB).47
If biobanks determine to engage in return, they will also need appropriate procedures and informatics capacity to deal with reidentification. Biobanks could approach reidentification in one of three ways. They could rely on primary researchers or collection sites to reidentify contributors when appropriate, they could themselves hold the key to the codes to reidentify contributors, or they could create an entity (such as a “trusted intermediary” or “honest broker”) to hold the key and perform reidentification.26,87 Note that if biobanks themselves hold the key to the codes (the second option), then the biobank would likely be conducting a research activity that would require an IRB-approved protocol and informed consent, unless the Common Rule criteria for waiver of consent are met. Yassin et al.87 elaborate on the option of using a “trusted intermediary,” saying that “The trustee’s role can be filled by a biospecimen resource, an entity within an academic institution not involved in the research, a subcontracted third party, or an informatics system.” Researchers are currently exploring automated informatics options to facilitate the return of IFs and IRRs, including the biobank using electronic interfaces to make detailed offers of data return so that contributors can elect what to receive.47,88,89
Finally, biobanks would need appropriate staffing or consultants, facilities, and funding to perform functions associated with deciding whether to return IFs and IRRs, and (if so) analyzing them and managing the return process. Staffing or consultant needs may include access to clinicians who are expert in analyzing the relevant findings.1 Facilities’ needs may include access to a CLIA-certified lab or labs to generate results appropriate for return.86 We elaborate below on all of these implications for biobanks.
Note that the precise allocation of responsibilities between primary research or collection sites and biobanks will vary by the design of the biobank research system. Suggesting that biobanks themselves should have some responsibilities for determining whether and how to return IFs and IRRs in a biobank research system does not mean that the biobank should have full responsibility and the primary researchers none. Coordination between primary researchers or collection sites and the biobank will be necessary, including coordination between their IRBs. As already noted, reidentification of contributor(s) may rest at the primary research site, biobank, at both, or with a “trusted intermediary.” Moreover, responsibilities for recontact may often best remain with the primary researcher or collection site. The primary researchers may, in some research designs, have ongoing contact with the contributor and thus be best suited to perform any recontact. However, recognizing that the biobank does bear some of the responsibilities for addressing IFs and IRRs in the biobank research system and for performing some of the specific functions involved has the potential to advance consistency, reliable performance of duties, and accountability to individual contributors and research participant populations. We also suggest strategies that biobanks can undertake cooperatively to build databases on return of IFs and IRRs, pool policies and procedures on return of IFs and IRRs, and promote rapid progress in developing sound approaches.
What is the content of those responsibilities? Four issues and who should address them
Having argued that some IFs and IRRs should indeed be considered for return, we next address the specific responsibilities involved. Identifying the core responsibilities will allow us to consider who in the complex system of biobank research is best positioned to discharge them and how. Four key responsibilities are:
-
1
Clarifying the criteria to determine what kind of findings are returnable, and the roster of returnable IFs and IRRs
-
2
Analyzing a particular finding to decide whether it should be offered to the individual contributor
-
3
Reidentifying that contributor (or contributors, if more than one is affected by the finding)
-
4
Recontacting the contributor(s) to offer the finding and genetic or other appropriate counseling
We refer to these four key responsibilities using the acronym CARR.
Step 1: Clarifying the criteria and roster. Early work on return of IFs and IRRs commonly did not separate this step from analyzing a particular finding for potential return. However, the major recommendations that have emerged thus far on the return of IFs and IRRs are framed in terms of general criteria for return rather than a specific list of which genetic, genomic, and other findings should be returned.1,75,86,90 There are good reasons for this. Principled resolution of the IFs and IRRs issues requires first some consensus on what general kinds of findings are appropriate for return. Because recommended criteria for return commonly focus on questions such as whether the meaning of the finding is established and its health or reproductive implications understood, whether the health or reproductive implications of the finding are significant, and whether return is “actionable” and thus can benefit the contributor, the specific genetic, genomic, and other findings that qualify will change with increasing knowledge. Thus, Fabsitz et al.86 urge the creation of a central advisory committee that can offer advice on what genetic and genomic findings are sufficiently well understood, significant, and actionable to qualify for return. Yet those authors are careful to recommend that such a body be advisory. There may be good grounds to adjust the committee-recommended criteria and roster of returnable findings according to the specific nature of the study or biobank, the study or biobank population, and their preferences for return. Indeed, Fabsitz et al. recommend the involvement of the research participant community, when identifiable, in addressing the return of IRRs.
Table 4 updates the prior list of major US consensus recommendations on criteria for return in Wolf et al.1 It adds our prior project’s 2008 recommendations and those subsequent. It also removes those not focused directly on return of IFs and IRRs or now superseded. As Table 4 suggests, two separate but interrelated issues are emerging: what criteria should ground an obligation or option to return and who should formulate those criteria, revise them over time with refinement of genetic and genomic knowledge, and recommend a roster of returnable results. On criteria, there is wide agreement that findings should be analytically valid and return should comply with applicable law. The requirement of analytic validity assures that the test results accurately reveal whether a particular genotype is present.91 Compliance with applicable law means that the finding was generated in compliance with CLIA.92 CLIA and its accompanying regulations impose a number of test quality assurance requirements when test results are being generated and returned for clinical purposes, including measures to ensure that the test result indeed belongs to the individual and has not been mixed up.93,94 Where the initial lab generating the findings is CLIA-certified or the researchers can reconfirm the results in a lab that is CLIA-certified, this is not an obstacle to return. (Note that this may require that researchers plan ahead to retain samples adequate for retesting in a CLIA-certified lab.) However, controversy surrounds the question of whether results can be returned if confirmation by a CLIA-certified lab is not available.86 The literature explores models that would allow researchers to provide or access testing in CLIA-certified labs.95 Fully clarifying best practices may require working with the CMS, the agency that administers CLIA.
A second general criterion garnering significant, though not complete, agreement is that the contributor or research participant has consented to receipt of this information.76,86,96,97 Ideally researchers and IRBs will anticipate the question of whether to return IFs and IRRs before data and samples are initially collected and the collecting researchers will ask contributors whether they wish to be recontacted in the future to receive such findings. Contributors may also be asked what kinds of IFs and IRRs they wish to receive, depending on the major categories expected in anticipated research.86,98 Debate surrounds the question of whether researchers should or may return results when the contributor has not given earlier consent to return or has earlier refused return, if the finding has high health importance and is clinically actionable (e.g., when return of a finding of a genetic variant associated with catastrophic reaction to a commonly used drug would allow avoidance of that drug).86 This question is hard to answer in the abstract and may require consideration in the context of the particular biobank, study population, and study type in question, ideally with the involvement of representatives from the study population and research on contributor preferences for return, as discussed in more detail below.
Another consent-related debate is whether to abstain from offering the return of IFs and IRRs or limit the return to only the most important findings when the individual contributors gave no consent for return because they gave no consent for research at all. As noted above, some biobank research and secondary research on data and/or samples from biobanks are conducted without consent, as the data and samples used were collected in clinical care and deidentified before biobank and secondary research, so the research is not considered human subjects research. In these research designs, the contributor has not consented to the research and thus has not consented to the return of IFs or IRRs. How to handle return in this scenario is another hard question. Options include refraining from any return, returning only the most important and clinically actionable findings, or contacting contributors to seek consent for return. The recent DHHS Advance Notice of Proposed Rulemaking2 raises the possibility that in the future contributors will be asked for consent to research on data and samples collected in clinical care, and thus could be asked at that point whether they wish to be recontacted in the future about findings of potential clinical importance.
Progressing beyond consent, analytic validity, and compliance with applicable legal requirements, we reach the question of what findings should or may be offered back to contributors. Here, recommendations vary. Our prior project’s recommendations article1 and the more recent paper by Fabsitz et al.86 differentiate those findings that should be offered, those that may be offered, and (in Wolf et al.1 plus Berg et al.99) those that should not be offered, recognizing three categories for IFs encountered in clinical practice and public health. We recommend below what should generally fall in each of these three categories (should offer, may offer, and should not offer). However, this is really the starting point for a biobank research system, which may further modify these criteria in a way that is sensitive to the biobank population and biobank research design.
We suggest that those IFs and IRRs that should be offered back to contributors are those of high importance and likely net benefit to the contributor. These include findings revealing substantial risk of a serious health condition, a criterion that addresses both the likelihood of developing the condition and the seriousness of the condition. Note that this criterion for return is more inclusive than the “should disclose” criterion stated in our earlier project’s recommendations, which required “significant risk of a condition likely to be life-threatening [or] grave.”1 Our revision strives to establish more consistency with the subsequent group recommendations from Fabsitz et al.86 under NHLBI auspices (“important health implications…and the associated risks are established and substantial”). This broadening of the criterion for “should be offered back” prompted debate in our author group. Some would continue to restrict “should offer back” to conditions that are life-threatening or grave. A significant majority of our group, however, supported moving to “substantial risk of a serious health condition,” in greater alignment with Fabsitz et al. In any case, the core question, as we suggested in our prior project’s article,1 is whether return offers strong net benefit from the contributor’s perspective. Evaluating net benefit from the contributor’s perspective can be done in several ways: by asking individual contributors, by conducting research on the contributor population to ascertain prevalent views, or by asking the biobank’s ICOB or other return of results committee (ideally in concert with a community engagement committee) to perform the evaluation as best they can from the standpoint of a reasonable contributor in that biobank research system.
Part of offering strong net benefit is actionability. This idea has appeared in multiple recommendations documents, but is variously understood. In keeping with Fabsitz et al., we suggest that the term should be understood broadly and, again, from the contributor’s perspective. The core question is whether return of the IF or IRR offers the contributor and/or the contributor’s clinician the option to take action with significant potential to alter the onset or course of disease, such as by allowing heightened surveillance, preventive actions, early diagnosis, or treatment options. Although some commentators might understand actionability narrowly from the clinician’s perspective (can we prevent or cure?), we instead define actionability from the perspective of the individual facing the risk and potential disease (can I and/or my clinician take action to prevent or alter the course of my condition or to tailor my treatment?).
In our earlier project’s paper, we included some findings of reproductive importance among those that should be offered back to contributor.1 These were findings indicating that offspring would bear a substantial risk of a serious health condition, when the finding could be used to ameliorate the condition or avoid the risk. Including reproductive importance among the criteria warranting return of IFs and IRRs is more controversial than restricting “should return” to findings bearing on the individual’s own health risk. The subsequent article by Fabsitz et al. treats reproductive importance as a criterion for discretionary return rather than recommended return (the researcher “may” return, rather than “should” return). Their article similarly treats personal meaning or utility as an occasion for discretionary return.
Thus, both Wolf et al.1 and Fabsitz et al.86 recognize reproductive importance as legitimate grounds for return of IFs and IRRs. This makes sense in genetic and genomic research, where some of the key findings concern heritability. We here follow the path of the Fabsitz et al. paper by suggesting that findings of reproductive importance should fall in the “may return” category. Our group debated whether a subset of reproductive findings belong in the “should return” category, but did not reach agreement on this point. Thus, return of reproductive findings, together with findings of personal utility (i.e., useful for life planning and decisions), should be discretionary.
A number of biobank characteristics can make return of IFs or IRRs more difficult for the biobank and potentially less beneficial to the contributor. Aggregating a large quantity of data or samples from a large number of contributors means that any policy on return will be more costly and labor-intensive than when undertaken in the context of a smaller-scale primary research study. Efforts are under way to estimate the number of genetic findings that might warrant consideration for return.100 Additionally, because biobanks archive data and samples for long periods of time and multiple research uses, a significant amount of time may elapse between collection of the data or samples and identification of an IF or IRR, potentially making relocating and recontacting the contributor challenging if not impossible and reducing the utility for the contributor (as predicted disease or death, for example, may have already occurred). Biobanks typically take steps to deidentify data and samples before they are released to secondary researchers for analysis (requiring de-identification at the collection site or doing it at the biobank or depending on a third entity (sometimes referred to as a “trusted intermediary” or “honest broker”) to do this), and may take further affirmative steps to prevent reidentification (as urged, for example, by Brothers and Clayton16).
The greater difficulty and cost of biobank return, the lower likelihood of benefit with lapse of time, and the reality that some contributors will not have consented to research, justify more restrictive criteria for return in biobank research than primary research. We recommend that the category of findings warranting an affirmative duty to return (“should return”) be limited to findings of high health importance to the contributor, while discretionary return (“may return”) should apply to findings of lesser health importance and those of reproductive importance and personal utility.
Translating criteria into a roster of returnable IFs and IRRs that trigger a responsibility to return or the option of discretionary return is an effort with the potential to simplify the ethical task of deciding whether to offer IFs and IRRs and create some consistency across biobanks and studies. As noted above, some commentators have suggested creating a Central Advisory Body to assemble such a roster nationally and refine it over time as new genetic and genomic tests are understood, their meaning established, and their analytic validity determined.86 We too endorse this recommendation, but note the limits of such an effort. The roster envisioned is a starting point, not an end-point, for ethics deliberation tailored to individual biobanks and study populations. Important research is under way and will continue on what findings different study populations and individuals regard as valuable for their own health, reproductive decision-making, and personal life (and death) planning.
Moreover, most efforts that have been suggested and are being piloted to assemble a roster of returnable results in genetic and genomic research have focused only on genetic and genomic results.101 Yet IFs and IRRs generated by biobank and secondary research will be broader and include nongenetic findings. IFs uncovered in ascertaining the suitability of an individual for participation in the biobank research and in baseline screening are likely to be nongenetic.102 Thus, the website for UK Biobank promises return of enrollment and baseline screening IFs such as elevated blood pressure.22,23 Even after biobank research itself has begun, biobanks and secondary researchers may well find nongenetic IFs, such as discrepant diagnosis when tumor specimens are reanalyzed at the biobank24or unrecognized phenotypic findings of significance in the electronic medical records, when those records are linked to genetic analyses for the purposes of research, as at eMERGE sites.9 Further, some biobanks are linking genetic and nongenetic data, such as neuroimaging scans, to look for connections between individuals’ genetic make-up and brain abnormalities;103,104 such linked databases may generate nongenetic IFs and IRRs for potential return from those neuroimaging scans. Finally, a number of biobanks are collecting information on environmental exposures.9,105 These data sources may generate IFs and IRRs for potential return as well, such as discovery of a previously unrecognized exposure of acute health importance and actionability.
Thus, any central roster of returnable genetic and genomic results will have important but confined utility, as it will not provide guidance on nongenetic findings. In addition, it will be difficult if not impossible to anticipate all of the IFs that may arise in various biobanks and studies. That said, we would recommend that any central body assembled to give advice on returnable IFs and IRRs also serve as a clearinghouse for policies and consensus statements on the returnability of both genetic and nongenetic IFs/IRRs. In our prior project’s consensus article, we addressed nongenetic IFs in neuroimaging and computed tomography (CT) colonography research, as well as genetic IFs.1 In addition, we recognized influential classification schemes already published for IFs in imaging research. Such classification schemes should be collected, made centrally and readily available, and compared (as we did in our prior IFs project) to glean cross-cutting lessons for improvement and harmonization. In addition, the central authority could support the creation of a central database to report and study IFs and IRRs across biobanks and studies. One example of a fledgling data source posted on the web is the Incidental Findings Files posted by GENEVA,101 a GWAS program under the aegis of the NIH-wide Genes, Environment and Health Initiative.
In addition to the need for a central source of advice and information, biobanks themselves and networks or consortia of biobanks (such as the eMERGE Network) need the capacity to address issues involving IFs and IRRs and to tailor central advice on criteria for return to the realities of the biobank’s contributor population, roster of studies, degree and type of deidentification, and capacity for recontact. Increasingly we see biobanks and networks creating such a committee. Use of an ICOB,47 for example, has been adopted by the Coriell Personalized Medicine Collaborative.106 The eMERGE Network has a Return of Results Oversight Committee.9,107 GENEVA has a Committee on Incidental Findings.108 Mayo Clinic has established a Biospecimen Trust Oversight Group to determine what results to return and the method for doing so.109 Other biobanks and networks may assign consideration of criteria for returnability to their IRB or conceivably their DAC. Some biobanks will find it useful to have all three entities, with the IFs/IRRs committee focusing on that issue, the IRB integrating those concerns with broader human subjects concerns, and the DAC addressing the question of how secondary researchers seeking to access biobank data and samples should handle IFs/IRRs and report them back to the biobank. Indeed, biobanks should consider writing into their contractual agreements with collection sites on one end of the process and secondary researchers on the other end (i.e., into collection agreements, MTAs, and DAAs or DUAs) provisions on how IFs and IRRs will be addressed. We see evidence of this already. For example, the Framingham Heart Study provides in its secondary (or “ancillary”) research application procedures that the secondary researcher must anticipate possible clinical findings and plan for the return of results to participants: “The proposing investigator must clearly delineate any findings of clinical significance that may result from the study, including genetic findings, and propose how these will be handled, including reporting to participants and their physicians and providing recommendations for follow-up. This includes IFs, such as pathology identified from an imaging study that is not the focus of the study.”110
Step 2: Analyzing a particular finding. If the first step is identifying the criteria to be used in determining whether findings should or may be offered back to contributors, the next step is considering a particular finding to determine whether it meets those criteria. This will involve consideration of a particular finding in the context of a specific study population or subpopulation. At this step, researchers and biobanks will ask questions such as “Is this incidental finding of BRCA2 in a study of pancreatic cancer genetics appropriate for return?”
Several questions will routinely arise. First, if the finding (e.g., the BRCA2 variant) is already available to patients through clinical genetics testing, does this alone mean that researchers and biobanks do not have any duty to return it? We suggest that when the patient’s medical records reveal prior diagnosis then there is no duty to return the finding. However, in the absence of documented prior diagnosis, the theoretical availability of findings through genetic testing outside of the research context does not reduce the duties incumbent on researchers and biobanks to address and return IFs and IRRs. Findings that are clinically actionable are often actionable in a particular timeframe; refraining from returning the IF or IRR now because at some time in the future the contributor may (or may not) seek clinical genetic testing may significantly reduce the utility of the information or eliminate utility entirely. When researchers encounter IFs or IRRs, it is often the case that theoretically this same finding could be produced in a clinical setting. This is especially true of IFs, which are, by definition, not the object of study. If the hypothetical availability of this information in a clinical context obviated the duty to return, there would be little if any duty to return any finding. It is precisely because in reality the researcher has a piece of information that may be new to the individual that the duty to return even arises.
Another question that will routinely arise is whether a particular finding is well-enough understood and established to return. This is an area of controversy. Some recommendations on the return of IFs and IRRs require that findings have “established risk.”86 However, the Personal Genome Project, although treating some findings as not ready for return, is nonetheless exploring the option of return with information sheets that indicate the level of scientific confirmation, rather than treating the degree to which a finding is established as a binary yes/no matter.111 Although the purpose of having a central authority clarify the roster of returnable findings in Step 1 is partly to identify those findings sufficiently established to warrant return, there will inevitably be some findings discovered that are not on that roster and for which there is not a well-established literature, that nonetheless raise serious concern (e.g., a deletion whose size and location raise high suspicion of strong clinical importance). These must be addressed on a case-by-case basis.
Finally, deciding whether to return a particular finding in light of the general criteria for return will again require addressing the question of whether the finding was produced in a CLIA-certified lab. If not, reconfirmation in a CLIA-certified lab is warranted, as noted above.
In determining who should perform the function of analyzing a particular finding in light of the general criteria and recommended roster of returnable IFs and IRRs, we again turn to the committees that biobanks and biobank networks are developing—ICOBs, Return of Results Committees, and Incidental Findings Committees. Concentrating this function in a committee that can learn from experience and build expertise over time makes sense. Including in that committee not only expertise on the scientific data in question (genetic and nongenetic), but also clinical expertise from professionals such as genetic counselors, ethics expertise, and representation from the biobank contributor population allows robust analysis of the values questions involved.
In proposing that this function be discharged at the level of the biobank or biobank network, we are suggesting that this will generally be preferable to leaving this function to collection sites or multiple secondary researcher sites. By locating at the biobank level the Step 1 responsibility of developing general criteria for return as well as the Step 2 responsibility for evaluating a particular finding, one can maximize uniformity of decision-making about IFs and IRRs across the whole three-stage process of biobank research. Biobanks are further in a position to use their bargaining power and contractual documents with collection sites and secondary researchers to require that IFs and IRRs for biobank contributors go through the biobank committee or processes using biobank-accepted criteria. Biobanks may also be best positioned to fund this function and sustain it over the prolonged period of use of the data and samples. Finally, biobanks are probably best positioned to document how they are handling IFs and IRRs, feed that into a database, conduct research to improve this process over time, and engage with the biobank contributor population to address and improve handling of IFs and IRRs in a way that is transparent, accountable, and comports with contributor population values regarding return.
Step 3: Reidentifying the contributor(s). Return of IFs and IRRs will generally require reidentification of the contributor individual(s). Biobanks vary in the amount of identifying information, if any, that reaches the biobank itself. Different biobanks within the eMERGE Network, for example, approach this differently. Vanderbilt’s BioVU robustly deidentifies samples using a process of generating a unique identifier that guards against ready reidentification.9 On the other hand, Northwestern’s NUgene retains the capacity to reidentify participants.112 Mayo’s biobank is making this a focal topic of community engagement: “Since genomic data cannot easily or with certainty be fully de-identified or anonymized, an important aim of the Mayo eMERGE project is to engage extensively with research participants and the community regarding best practices….”9
Variation in degrees of deidentification and who holds the key to the code for reidentification is a challenge for return of IFs and IRRs from biobank research. When only the primary researcher (or collection site) holds the key, the capacity to return IFs and IRRs may be limited by research funding at the primary research (or collection) site, whether the original investigator or someone else capable of performing reidentification remains there, whether the key for reidentification can be found and used for reidentification even after a potentially significant lapse of time, and the policy of that particular IRB on reidentification and return of results. This will likely produce multiple different outcomes within a single biobank research system when the biobank contributor population derives from multiple primary research or collection sites. It also means that the particulars of individual researchers and institutions will determine how the system discharges ethical responsibilities. We recommend the development of approaches to reidentification that allow greater consistency and predictability within a biobank research system. For biobanks that currently conduct research on identified data and/or samples or themselves hold the key and can readily reidentify contributors, it is no stretch to say they bear responsibilities for reidentification. Under the Common Rule, they are conducting human subjects research and assigning them the responsibility for reidentification to return the IFs and IRRs deemed important to return does not change that. However, for biobanks that do not hold the key and cannot readily reidentify contributors, it is a different matter. Such biobanks are not considered to be conducting human subjects research under the Common Rule. Asking them to hold the key themselves would change that. However, when reidentification and recontact is indeed possible, at least at the primary research or collection site, an alternative to having the biobank itself hold the key is to create an independent, “trusted intermediary” to hold it.87,113 This means that after ascertaining in Steps 1 and 2 that an IF or IRR worthy of return has been found, the reidentification responsibility would rest on the “trusted intermediary.” In the absence of such an entity, it would need to rest at the initial collection site, as in what we have called the “conventional view.”
Step 4: Recontacting the contributor to offer the finding. Once an IF or IRR appropriate for return has been found and the contributor individual(s) reidentified, we reach the questions of who should recontact the contributor to offer the finding and how. In our prior project’s article,1 we urged that the return be conducted by an individual with the expertise to answer questions about the finding being returned. This may require a genetic counselor for genetic or genomic findings and another clinician for some other findings (such as an oncologist for cancer-related findings). At the same time, we recognized a limit to this return function. The goal was not to supply clinical care or a clinical work-up of the finding, but to put the participant in the position to make an informed decision about what next steps to take, including seeking clinical work-up and care. We urged that the professional conducting the recontact be prepared to suggest clinicians for further pursuit of the finding, including for participants without health-care insurance or other ready access to clinical care.
A further question is to whom the finding should be communicated—the contributor or the contributor’s primary care physician (PCP). In our prior project’s paper, we argued that offering the return of findings to the research participant was ethically preferable to insisting on return to the PCP.1 We suggested that many people did not have an established PCP, and that individuals may wish to protect their privacy by deciding what clinician to consult and where the information should be recorded. However, there was debate in the follow-on project presented here about the merits of return directly to contributors versus to their PCP. An available compromise is to ask contributors whether they wish to receive information about such findings themselves or would prefer to receive the information through their PCP or another clinician whom they can identify.
In the biobank context, there is an additional important question of whether the biobank itself should recontact the contributor or this is best left to the primary research or collection site. Biobanks will vary in whether the biobank itself has any history of contact with the contributor and any ongoing relationship, or whether a separate primary research or collection site (such as a hospital or health-care setting) is the entity with such a relationship. This makes it difficult to offer a uniform recommendation on who should conduct the recontact. Deciding who should recontact the contributor should involve considering who has a history of contact with the contributor and who has the resources to provide an appropriate professional to conduct recontact. The questions of who should perform recontact and how should be anticipated in designing the biobank and its system of obtaining data and samples from primary researchers or collection sites. The agreements governing the collection and flow of data and samples to the biobank should address this recontact question, so it is clear who bears this responsibility.
On the other side of the biobank flow chart ( Figure 1 ), it is difficult to imagine a circumstance in which secondary researchers should be the ones to recontact a contributor, even when it is their research that uncovers the IF or IRR. We are unaware of a model in which researchers conducting secondary reanalysis have any contact with the contributors. Indeed, MTAs and DAAs commonly forbid secondary researchers from even attempting reidentification of contributors.80 Under our recommendations, IFs and IRRs discovered by secondary researchers should flow back to the biobank for analysis and handling. Thus, recontact is a matter for the biobank or primary research (or collection) sites to handle, not secondary researchers.
The “how” of recontact and method of reporting IFs and IRRs are subjects of active research. Kohane et al.47 recommend a computer interface to offer findings, governed by ICOB determination of returnable findings and contributors’ individual expressions of preference for certain kinds of findings through the interface. Roberts et al.114 have offered research participants a choice between in-person and telephone genetic counseling to reach those who have permanently or seasonally relocated. The Coriell Personalized Medicine Collaborative has developed a methodology and format for return, differentiating risks based on genetic and nongenetic factors.106 Both the REVEAL Study (focusing on apolipoprotein E and the risk of Alzheimer disease) and ClinSeq (focusing on atherosclerotic disease) have developed means for reporting individual findings as well.115,116 In all of these cases, what is offered back to contributors (or research participants) are not raw data, but rather interpreted findings that have been selected as appropriate for return.
Figure 2 summarizes the roles and responsibilities described above for the return of IFs and IRRs through the four-step CARR process. Part III then distills this and the discussion above into 10 concrete recommendations.
Part III: Crystallizing 10 Recommendations
This section synthesizes the roles and responsibilities suggested above to offer 10 concrete recommendations.
Recommendation 1
A primary researcher conducting the research and the researcher’s institution will be responsible for managing IFs and IRRs in that research. However, when the primary researcher is contributing data and/or samples to a biobank or like repository, the primary researcher (who may also represent a “collection site”), the biobank, and secondary researchers who access data and/or samples from the biobank are all part of a biobank research system. They each have a role to play in responsible management of IFs and IRRs as data and samples flow through the system. The biobank itself should strive to see that the biobank research system fulfills IF/IRR responsibilities, as the biobank is the central resource, with typically the longest continuous existence and funding (compared to primary and secondary researchers), contractual and operational relationships with both primary and secondary researchers, and recognized duties of custodianship, stewardship, and accountability.
Recommendation 2
Biobanks should strive to ensure that the process of designing a biobank research system includes development of explicit policy on whether IFs and IRRs will be returned. Some biobank research systems may be designed to irretrievably strip identifiers at the initial collection of data and samples and to take other steps to block reidentification. By blocking reidentification, this design will block the return of individual findings as well. However, many biobank research systems will retain a capacity to reidentify individual contributors—either at the primary research or collection site, at the biobank itself, or by a “trusted intermediary.” When the capacity to reidentify individual contributors remains, the biobank should work to create a responsible approach to return of IFs and IRRs within the biobank research system, in keeping with the recommendations below.
Recommendation 3
In a biobank research system, responsibilities for handling IFs and IRRs can be broken down into four steps organized by the acronym CARR:
-
1
Clarifying the criteria for evaluating findings and the roster of returnable IFs and IRRs
-
2
Analyzing a particular finding to decide whether it should be offered to the contributor
-
3
Reidentifying that contributor (or contributors, if multiple individuals one are affected by the finding); and
-
4
Recontacting the contributor to offer the finding.
To address the fulfillment of the CARR responsibilities in the biobank research system of which the biobank is a part, the biobank should have a multidisciplinary committee such as an Incidental Findings Committee, Return of Results Committee, or ICOB. These committees, focusing on IFs and IRRs issues, will need to interact with other relevant committees, particularly the IRBs at the biobank and at the primary research or collection sites.
Recommendation 4
In a biobank research system, the biobank should work with the primary researchers (or collection sites) and secondary researchers involved in the biobank system to clarify the criteria for evaluating findings (such as analytic validity, established and substantial risk of a serious health condition, and actionability) and roster of returnable IFs and IRRs. This roster should be reviewed and updated regularly in light of the literature, to incorporate new genetic and genomic findings. The biobank should consider the recommendations of a Central Advisory Body, if any. Creation of a Central Advisory Body to recommend a core roster would promote consistency among biobank research systems. However, biobanks will commonly need to consider whether the nature of their contributor population, their research collection, and the research facilitated suggests adjustments to the recommended roster.
-
(A) We recommend that researchers in the biobank research system should offer to return IFs and IRRs that meet all of the following criteria:
-
• The findings are analytically valid;
-
• Returning them to the contributor comports with applicable law, including CLIA (which may require ascertaining or verifying results in a CLIA-certified lab);
-
• The contributor has been offered the option of consenting to return of individual findings (either in the initial informed consent process, or in a subsequent consent process that may be a request for an individual’s consent or part of a larger effort to elicit many contributors’ consent) and has opted to receive them;
-
• The findings reveal an established and substantial risk of a serious health condition; and
-
• The findings are clinically actionable, meaning that return of findings of health importance allows the contributor or contributor’s clinician to take action with significant potential to prevent or alter the course of the condition or to alter its treatment.
-
(B) We recommend that researchers in the biobank research system may offer to return IFs and IRRs that fail to meet the above criteria, if they instead meet all of the following criteria:
-
• The findings are analytically valid;
-
• Returning them to the contributor comports with applicable law, including CLIA (which may require ascertaining or verifying results in a CLIA-certified lab);
-
• The contributor has been offered the option of consenting to return of individual findings (either in the initial informed consent process or subsequently, as indicated above in (A)) and has opted to receive them; and
-
• The findings reveal an established and substantial risk of likely health or reproductive importance or personal utility to the contributor and return is likely to provide net benefit from the contributor’s perspective.
-
(C) We recommend that researchers in the biobank research system not return IFs and IRRs offering unlikely net benefit from the contributor’s perspective, including findings whose likely health or reproductive importance or personal utility cannot be ascertained.
Recommendation 5
In the context of a biobank research system, biobanks should analyze whether a particular finding qualifies as an IF or IRR that should be offered to a consenting contributor or contributors. An exception to this is when the IF or IRR arises in the primary research and the primary research constitutes human subjects research overseen by an IRB, in which case the primary researcher and researcher’s institution should generally bear this responsibility. Collection agreements between biobanks and collection sites or primary researchers should specify the division of responsibility for handling IFs and IRRs, including analysis of particular findings.
Recommendation 6
In the context of a biobank research system, primary researchers (or collection sites) and biobanks should anticipate how they will handle identification or reidentification of contributors for return of IFs and IRRs. They should strive to reach agreement on this in advance, which is reflected in the collection agreement between the primary researcher (or collection site) and biobank. When data and/or samples are deposited in a biobank, either the biobank or a “trusted intermediary” (sometimes called an “honest broker”) may receive the key to codes (if any) to allow reidentification, rather than counting entirely on the primary research or collection site to maintain the capacity for reidentification over an extended period of time during which research is continuing.
Recommendation 7
In the context of a biobank research system, primary researchers (or collection sites) and biobanks should anticipate how they will handle recontacting contributors to offer return of IFs and IRRs. They should strive to reach agreement on this in advance, which is reflected in the consent form and the collection agreement between the primary researcher (or collection site) and biobank. In general, if the primary researchers (or collection site) have had direct contact with the contributor, and especially if they maintain an ongoing relationship with the contributor and have the capacity and expertise for recontact and communication of the findings, they should handle this. In other cases, the biobank should handle this, either directly or by engaging health professionals with appropriate expertise, such as genetic counselors. Findings should be returned in a form that is understandable to the contributor and useful to a physician or other clinician, such as a genetic counselor, with whom the contributor may choose to pursue the finding. This will generally mean that the finding should be returned both through direct conversation with the contributor and in writing, to minimize miscommunication and facilitate follow-up. Those professionals recontacting the contributor and communicating the findings should be prepared to offer clinical referrals, including for genetic counseling. If the contributor prefers that the finding be communicated directly to the contributor’s health professional, this preference should be honored.
Recommendation 8
Biobanks should strive to engage actively with contributors and potential contributors to clarify preferences and priorities of return of IFs and IRRs, including the roster of findings to be returned and the methods of return. Biobanks should facilitate the involvement of contributors and potential contributors in biobank policy-setting on this issue. Further research on the preferences, experiences, and outcomes of contributors and research participant populations with return of IFs and IRRs is needed. Researchers in biobank research systems and biobanks themselves have an important role to play in facilitating and conducting that research, to aid progress over time.
Recommendation 9
Researchers in a biobank research system should strive to communicate aggregate results not only through the scholarly literature, but also to the population of interested contributors using accessible media such as newsletters, e-mail, and other means of communication appropriate for the population. Offering links to the scholarly literature allows those who are interested to pursue reported findings in greater depth. Primary researchers and biobanks should consider in advance how they will respond to follow-up inquiries from individual contributors wishing to pursue the individual importance of aggregate findings.
Recommendation 10
Research and biobank funders and regulators have a crucial role to play in making sure that research and biobank budgets adequately support responsible management of IFs and IRRs. They also have a crucial role to play in making sure that research oversight and biobank guidelines support sound handling of IFs and IRRs. Further work is needed to articulate ethical standards for research with human data and/or samples that is not considered human subjects research under the Common Rule.
Part IV: Recommendations for New and Existing Biobanks
Fulfilling these 10 recommendations will pose different challenges for new biobanks that can address these recommendations prospectively than for biobanks already in existence, many of which were created before the current debate on returning IFs and IRRs arose.
New biobanks
New biobanks should address the IFs and IRRs issue prospectively before beginning operations. They should consider the 10 recommendations offered in design of the biobank research system, creation of governance structures and committees, drafting the governing documents (including biobank agreements with collection sites or primary researchers, and MTAs and DAAs or DUAs between the biobank and secondary researchers), staffing, and funding.
To be more specific, in structuring biobank interaction with primary researchers or collection sites, biobanks should make sure that primary researchers (or collection sites) specify how they plan to handle the issue of IFs and IRRs, and indicate that they have consulted their IRB in erecting this plan. Biobanks will need to establish an agreement with primary researchers (or collection sites) on the respective roles the biobank and primary research (or collection site) will play in the CARR process. Together they will need to consider whether key codes will be housed not just at the primary research (or collecting) institution but also at the biobank or a “trusted intermediary,” as discussed above. They will also need to consider how any consent process will address whether any IFs/IRRs identified in the biobank research system will be offered back to contributors, what type of findings will potentially be returned, whether individual contributors consent to return, and whether contributors are willing to share contact information and update that information over time to facilitate recontact.
In structuring the biobank’s own operations, the facility will need to establish policies, committees and advisory resources, procedures, staff, and funding to support fulfillment of the responsibilities suggested in Recommendations 1–10 above. Committees and other advisory resources should include a dedicated committee on return of IFs and IRRs (such as an ICOB), an IRB, a DAC to manage access by secondary researchers, and a means of community consultation such as a Community Engagement Committee. Some biobanks may be able to combine some of these functions into a smaller number of committees, but for large and complex biobank research systems, all may be needed. Biobanks will also need to consider informatics challenges posed by IFs and IRRs, including how to annotate data and samples to track the primary research institution (or collection site), how to note that the consent process addressed handling of IFs and IRRs, and how to track contributor consent to or refusal of recontact with respect to IFs and IRRs. Biobanks will further need to consider their use of or access to CLIA-certified labs to generate returnable findings. Importantly, biobanks will need to consider how to hold codes for reidentification or to establish a “trusted intermediary” to perform this function. They will also need to consider their access to clinical personnel (including genetic counselors) as needed to consult on IFs and IRRs and potentially perform recontact. Clinical personnel could usefully collaborate in advance to establish a methodology and format for return of IFs and IRRs, considering those already described in the literature.47,106
In structuring biobank interaction with secondary researchers, biobanks will need to establish DAC and ICOB (or other IF/IRR committee) policies to assure that secondary researchers take steps to facilitate sound handling of IFs and IRRs.20 Secondary researchers will need to address in their application for access to data and/or samples the potential of their research to generate IFs and IRRs that may be returnable. To facilitate this judgment, the secondary researchers will need to receive from the biobank a document specifying what IFs and IRRs are deemed returnable. The closer this document is to an actual roster of returnable findings, the easier it will be for secondary researchers to respond. Secondary researchers are already commonly asked to agree that they will refrain from reidentifying contributors. MTAs and DAAs or DUAs between the biobank and secondary researchers should reflect all of these understandings.
Preexisting biobanks
Retrofitting preexisting biobanks to cope with IFs and IRRs is more challenging. These biobanks vary in terms of the type of consent (if any) elicited from contributors. Some biobanks are comprised of data and/or samples collected without research consent (e.g., excess blood samples remaining after clinical laboratory analysis and then deidentified for research use). In these biobanks, contributors may not realize that their data or samples are being used in research. The literature debates whether recontact to elicit consent to return IFs and IRRs would be an unwelcome and inappropriate surprise.97,117 In this no-consent case, researchers or the biobank may nonetheless encounter findings of high health significance and actionability. Thus, an emerging recommendation is to allow some return, where possible, but only of the most important findings, that is, those addressed in Recommendation 4(A) above. In some biobanks, however, the biobank (ideally in coordination with the primary researcher) may find it feasible and ethically appropriate to recontact the population of contributors (or those with potentially returnable IFs or IRRs) to ask whether they consent to recontact and return. In those cases, return would not be so limited, as this would move the data and/or samples out of the category of “no consent to return IFs/IRRs.”
Other biobanks may aggregate data and/or samples collected with contributor consent to research, but the consent process did not address return of IFs and IRRs. To determine potential returnability of IFs/IRRs, the biobank (ideally in coordination with the primary researcher) should examine the consent process used and consents obtained to determine whether these consents are adequate to allow broader the return of IFs and IRRs than in the no-consent case above or, conversely, suggest that contributors were opting out of return. If the consent process and forms are simply silent on IFs/IRRs, return should be limited as in the no-consent case, unless the biobank (ideally in coordination with the primary researcher) finds it feasible and ethically appropriate to recontact the population of contributors or those with potentially returnable IFs or IRRs to determine whether they do actually consent to recontact and return.
Finally, there are biobanks that aggregate data and/or samples collected with contributor consent to research and the consent process did address return of IFs and IRRs. Our research suggests that many, though not all, of the biobanks that have addressed return of IFs and IRRs in their consent process have done so by stating that they will not return any IFs or IRRs.3 Some of these biobanks took this position before the last few years of scholarship, dialogue, and research on return of IFs and IRRs and development of approaches for return. Other biobanks may have taken this position more recently. Where the consent specifically stated that no IFs or IRRs would be returned and contributors agreed to research use of their data and/or samples on those terms, it is difficult to argue for return, unless the biobank (probably in coordination with the primary researcher) finds it feasible and ethically appropriate to recontact the population of contributors or those with potentially returnable IFs or IRRs to determine whether they actually do prefer recontact and return.
Conclusion
Addressing the return of IFs and IRRs in the context of genetic and genomic research involving biobanks requires going beyond the current literature on return of findings in individual research projects. Considering the entire biobank research system is essential, including the three major actors: primary researchers or collection sites, the biobank itself, and secondary researchers. All have the potential to generate returnable IFs and IRRs.
This article considers the ethical underpinning of responsibilities that devolve on each of the three major actors in a biobank research system to collaborate in handling IFs and IRRs. We then offer systemic analysis of how those responsibilities can best be discharged both in new biobank research systems considering these problems prospectively and in preexisting research systems. By articulating significant new responsibilities for the biobank itself, considering how all major responsibilities should be addressed within the biobank research system, and articulating 10 concrete recommendations for handling IFs and IRRs, this paper breaks new ground. In the context of 21st century genetics and genomics, with research powered by biobanks and large archived data sets, resolving how to handle IFs and IRRs is essential.
Appendix: Definitions of “Biobank” and Related Terms
Biobank: “A stored collection of genetic samples in the form of blood or tissue that can be linked with medical and genealogical or lifestyle information from a specific population, gathered using a process of generalized consent.”67
Biological Resource Centre: The term is used “to identify the whole range of biorepositories and related services dedicated to cancer research that are based on collections of human specimens…. The definition of a BRC not only involves the infrastructure for collection, archival and storage of biospecimens and data, but also the procedures and services for informing individuals who are approached to participate in a study, obtaining their consent, collecting and processing specimens for secure long-term storage, accessing and retrieving specimens appropriate for analysis, processing for preparation of biomaterials (e.g., DNA, RNA, proteins) for quality control, packaging and shipping specimens, etc.”118
Biorepository: “An organization, place, room, or container (a physical entity) where biospecimens are stored. In the context of the NCI Best Practices, only biorepositories containing human specimens collected with an intention to use them for research purposes (research biorepositories) are addressed. The physical structure, policies, and the biospecimens and data contained within it are defined collectively as a biospecimen resource….”26
Biospecimen Resource: “[A] collection of human specimens and associated data for research purposes, the physical entity where the collection is stored, and all relevant processes and policies. Biospecimen resources vary considerably, ranging from formal organizations to informal collections of materials in an individual researcher’s freezer....”26
Human Biobanks and Genetic Research Databases: “[S]tructured resources that can be used for the purpose of genetic research and which include: (a) human biological materials and/or information generated from the analysis of the same; and (b) extensive associated information.”77
Human Tissue Repositories: They “collect, store, and distribute human tissue materials for research purposes. Repository activities involve three components: (i) the collectors of tissue samples; (ii) the repository storage and data management center; and (iii) the recipient investigators”15 (emphasis omitted).
Repository: “An entity that receives, stores, processes and/or disseminates specimens, as needed. It encompasses the physical location as well as the full range of activities associated with its operation. It may also be referred to as a BIOREPOSITORY or BIOBANK.”70
Disclosure
R.H. is of counsel at Faegre Baker Daniels Law Firm and a shareholder in MR3 Medical, LLC. K.J.M. receives modest salary support as a consultant to the Mayo Clinic Biobank. The other authors declare no conflict of interest.

Roles and responsibilities for return of IFs and IRRs in a generic biobank research system (contributors not depicted). CAB, Central Advisory Body; DAA, Data Access Agreement; DAC, Data Access Committee; ICOB, Informed Cohort Oversight Board; IF, incidental finding; IRB, institutional review board; IRR, individual research result; MTA, Material Transfer Agreement.
References
Wolf SM, Lawrenz FP, Nelson CA,et al. Managing incidental findings in human subjects research: analysis and recommendations. J Law Med Ethics 2008;36:219–248.
US Department of Health and Human Services. Human subjects research protections: enhancing protections for research subjects and reducing burden, delay, and ambiguity for investigators. Federal Register 2011;76: 44512–44531. http://www.gpo.gov/fdsys/pkg/FR-2011-07-26/html/2011-18792.htm. Accessed 6 December 2011.
Johnson G, Lawrenz F, Thao M . An empirical examination of the management of return of individual research results and incidental findings in genomic biobanks. Genet Med, 2012;14:444–450.
Cambon-Thomsen A, Rial-Sebbag E, Knoppers BM . Trends in ethical and legal frameworks for the use of human biobanks. Eur Respir J 2007;30:373–382.
Johnston C, Kaye J . Does the UK Biobank have a legal obligation to feedback individual findings to participants? Med Law Rev 2004;12:239–267.
Winickoff DE . Partnership in U.K. Biobank: a third way for genomic property? J Law Med Ethics 2007;35:440–456.
Van Ness B, Ramos C, Haznadar M,et al. Genomic variation in myeloma: design, content, and initial application of the Bank On A Cure SNP Panel to detect associations with progression-free survival. BMC Med 2008;6:26.
Clayton EW, Smith M, Fullerton SM,et al. Confronting real time ethical, legal, and social issues in the Electronic Medical Records and Genomics (eMERGE) Consortium. Genet Med 2010;12:616–620.
McCarty C, Chisholm RL, Chute CG, et al. The eMERGE Network: a consortium of biorepositories linked to electronic medical records data for conducting genomic studies. BMC Med Genomics 2011;4:13.
International Myeloma Foundation. Bank On A Cure. http://myeloma.org/ArticlePage.action?articleId=670. Accessed 5 April 2011.
US Department of Health and Human Services. Protection of human subjects. 45 C.F.R. part 46.
Framingham Heart Study. About the Framingham Heart Study. http://www.framinghamheartstudy.org/about/index.html. Accessed 22 March 2011.
Office for Human Research Protection. Guidance on Research Involving Coded Private Information or Biological Specimens, 16 October 2008. http://www.hhs.gov/ohrp/policy/cdebiol.html. Accessed 6 December 2011.
Office for Human Research Protections. Guidance on Engagement of Institutions in Human Subjects Research, 16 October, 2008. http://www.hhs.gov/ohrp/policy/engage08.html. Accessed 5 February 2011.
Office for Protection from Research Risks. Issues to Consider in the Research Use of Stored Data or Tissues, 7 November 1997. http://www.hhs.gov/ohrp/policy/reposit.html. Accessed 6 December 2011.
Brothers KB, Clayton EW . “Human non-subjects research”: privacy and compliance. Am J Bioeth 2010;10(9):15–17.
Brothers KB . Biobanking in pediatrics: the human nonsubjects approach. Per Med 2011;8:79.
Cho MK . Understanding incidental findings in the context of genetics and genomics. J Law Med Ethics 2008;36:280–285.
Beskow LM, Burke W . Offering individual genetic research results: context matters. Sci Transl Med 2010;2:38cm20.
Dressler LG . Biobanking and disclosure of research results: addressing the tension between professional boundaries and moral intuition. In: Solbakk JH, Holm S, Hofmann B (eds). The Ethics of Research Biobanking. Springer: New York, 2009:85–99.
Yale University Institutional Review Boards. IRB Policy 720 Findings With Possible Health and Safety Significance for Research Participants, 13 January 2010. http://www.yale.edu/hrpp/resources/docs/IRBPolicy720Incidentalfindings_000.pdf. Accessed 20 February 2011.
UK Biobank. Your Questions Answered: Test Results. http://www.ukbiobank.ac.uk/faqs.php. Accessed 16 March 2011.
UK Biobank Ethics and Governance Framework, version 3.0, October 2007. http://www.ukbiobank.ac.uk/wp-content/uploads/2011/05/EGF20082.pdf. Accessed 17 January 2012.
NCI, Office of Biorepositories and Biospecimen Research. Workshop on Release of Research Results to Participants in Biospecimen Studies, Bethesda, Maryland, 8–9 July 2010, Workshop Summary, 4 March 2011. http://biospecimens.cancer.gov/global/pdfs/NCI_Return_Research_Results_Summary_Final-508.pdf. Accessed 6 December 2011.
Illes J, Kirschen MP, Edwards E,et al. Ethics. Incidental findings in brain imaging research. Science 2006;311:783–784.
NCI, Office of Biorepositories and Biospecimen Research. NCI Best Practices for Biospecimen Resources, 2011. http://biospecimens.cancer.gov/bestpractices/2011-NCIBestPractices.pdf. Accessed 10 January 2012.
Wallace SE, Kent A . Population biobanks and returning individual research results: mission impossible or new directions? Hum Genet 2011;130:393–401.
NIH Points to Consider for IRBs and Institutions in their Review of Data Submission Plans for Institutional Certifications under NIH’s Policy for Sharing of Data Obtained in NIH Supported or Conducted Genome-Wide Association Studies (GWAS), 31 May 2011. http://gwas.nih.gov/pdf/PTC_for_IRBs_and_Institutions_revised5-31-22.pdf. Accessed 10 January 2012.
Murphy J, Scott J, Kaufman D, Geller G, LeRoy L, Hudson K . Public expectations for return of results from large-cohort genetic research. Am J Bioeth 2008;8(11):36–43.
Clayton EW, Ross LF . Implications of disclosing individual results of clinical research. JAMA 2006;295:37; author reply 37–38.
Ormond KE, Smith ME, Wolf WA . The views of participants in DNA biobanks. Stanford J Law Sci Policy 2010;1:80–87.
Partridge AH, Winer EP . Informing clinical trial participants about study results. JAMA 2002;288:363–365.
Wendler D, Emanuel E . The debate over research on stored biological samples: what do sources think? Arch Intern Med 2002;162:1457–1462.
Beskow LM, Dean E . Informed consent for biorepositories: assessing prospective participants’ understanding and opinions. Cancer Epidemiol Biomarkers Prev 2008;17:1440–1451.
Kaufman D, Geller G, Leroy L, Murphy J, Scott J, Hudson K . Ethical implications of including children in a large biobank for genetic-epidemiologic research: a qualitative study of public opinion. Am J Med Genet C Semin Med Genet 2008;148C:31–39.
Beskow LM, Smolek SJ . Prospective biorepository participants’ perspectives on access to research results. J Empir Res Hum Res Ethics 2009;4:99–111.
Nicol D, Critchley C . Benefit sharing and biobanking in Australia. Public Understand Sci 2011; e-pub ahead of print 21 April 2011.
Pulley J, Clayton E, Bernard GR, Roden DM, Masys DR . Principles of human subjects protections applied in an opt-out, de-identified biobank. Clin Transl Sci 2010;3:42–48.
Vanderbilt BioVU. BioVU: Vanderbilt’s DNA Databank. http://dbmi.mc.vanderbilt.edu/research/dnadatabank.html. Accessed 23 March 2011.
Loukides G, Denny JC, Malin B . The disclosure of diagnosis codes can breach research participants’ privacy. J Am Med Inform Assoc 2010;17:322–327.
US Food and Drug Administration. Human research protections. 21 C.F.R. parts 50, 56.
Office for Human Research Protections. Regulations. http://hhs.gov/ohrp/humansubjects/index.html. Accessed 22 February 2012.
Wolf SM, Paradise J, Caga-anan C . The law of incidental findings in human subjects research: establishing researchers’ duties. J Law Med Ethics 2008;36:361–383.
Richardson HS, Belsky L . The ancillary-care responsibilities of medical researchers. An ethical framework for thinking about the clinical care that researchers owe their subjects. Hastings Cent Rep 2004;34(1):25–33.
Richardson HS . Incidental findings and ancillary-care obligations. J Law Med Ethics 2008;36:256–270.
Miller FG, Mello MM, Joffe S . Incidental findings in human subjects research: what do investigators owe research participants? J Law Med Ethics 2008;36:271–279.
Kohane IS, Mandl KD, Taylor PL, Holm IA, Nigrin DJ, Kunkel LM . Medicine. Reestablishing the researcher-patient compact. Science 2007;316:836–837.
HUGO Ethical, Legal, and Social Issues Committee. Statement on the Principled Conduct of Genetics Research, 1995. http://www.hugo-international.org/img/statment%20on%20the%20principled%20conduct%20of%20genetics%20research.pdf. Accessed 4 April 2011.
Knoppers BM, Joly Y, Simard J, Durocher F . The emergence of an ethical duty to disclose genetic research results: international perspectives. Eur J Hum Genet 2006;14:1170–1178.
Zawati MH, Van Ness B, Knoppers BM . Incidental findings in genomic research: a review of international norms. GenEdit 2011;9(1):1–8.
Council for International Organizers of Medical Sciences. International Ethical Guidelines for Biomedical Research Involving Human Subjects, 2002. www.cioms.ch/publications/layout_guide2002.pdf. Accessed 6 December 2011.
Clayton EW, McGuire AL . The legal risks of returning results of genomics research. Genet Med, 2012;14:473–477.
Fernandez CV, Kodish E, Weijer C . Informing study participants of research results: an ethical imperative. IRB 2003;25(1):12–19.
Shalowitz DI, Miller FG . Disclosing individual results of clinical research: implications of respect for participants. JAMA 2005;294:737–740.
Fernandez CV . Public expectations for return of results: time to stop being paternalistic? Am J Bioethics 2008;8:46–48.
Affleck P . Is it ethical to deny genetic research participants individualised results? J Med Ethics 2009;35:209–213.
Dressler LG . Disclosure of research results from cancer genomic studies: state of the science. Clin Cancer Res 2009;15:4270–4276.
Bredenoord AL, Kroes HY, Cuppen E, Parker M, van Delden JJ . Disclosure of individual genetic data to research participants: the debate reconsidered. Trends Genet 2011;27:41–47.
Chadwick R, Berg K . Solidarity and equity: new ethical framework for genetic databases. Nature Rev Genet 2001;2:318–321.
Chadwick R, Cutter M . The impact of biobanks on ethical frameworks. In: Hayry M, Chadwick R, Arnason V, Arnason J (eds). The Ethics and Governance of Human Genetic Databases: European Perspectives. Cambridge University Press: New York, 2007:219–226.
HUGO Ethics Committee. Statement on Benefit-sharing, 2000. http://www.hugo-inte--rnational.org/img/benefit_sharing_2000.pdf. Accessed 18 March 2011.
HUGO Ethics Committee. Statement on Human Genomic Databases, 2002. http://www.hugo-international.org/img/genomic_2002.pdf. Accessed 16 March 2011.
Wilson S, Chadwick R . Pursuing equality: questions of social justice and population genomics. In Hayry M, Chadwick R, Arnason V, Arnason J (eds). The Ethics and Governance of Human Genetic Databases: European Perspectives. Cambridge University Press: New York, 2007: 150–158.
Rynning E . Legal challenges and strategies in the regulation of research biobanking. In: Solbakk JH, Holm S, Hofmann B (eds). The Ethics of Research Biobanking. Springer: New York, 2009:277–313.
Chalmers D . Genetic research and biobanks. In: Dillner J (ed). Methods in Biobanking. Humana Press: Totowa, NJ, 2011:1–37.
Kaye J . Abandoning informed consent: the case of genetic research in population collections. In: Tutton R, Corrigan O (eds). Genetic Databases: Socio-ethical Issues in the Collection and Use of DNA. Routledge: London, 2004:117–138.
Secretary’s Advisory Committee on Genetics, Health, and Society. Policy Issues Associated With Undertaking a New Large U.S. Population Cohort Study of Genes, Environment, and Disease, 2007. http://oba.od.nih.gov/oba/sacghs/reports/SACGHS_LPS_report.pdf. Accessed 1 September 2010.
Wallace S, Bedard K, Kent A, Knoppers BM . Governance mechanisms and population biobanks: building a framework for trust. GenEdit 2008;6(2):1–11.
Winickoff DE . From benefit sharing to power sharing: partnership governance in population genomics research. In: Kaye J, Stranger M (eds). Principles and Practice in Biobank Governance. Ashgate Publishing: Burlington, VT, 2009:53–66. http://escholarship.org/uc/item/845393hh. Accessed 12 October 2010.
International Society for Biological and Environmental Repositories. 2008 best practices for repositories, collection, storage, retrieval and distribution of biological materials for research. Cell Preserv Technol 2008;6: 3–58.
Jeffers BR . Human biological materials in research: ethical issues and the role of stewardship in minimizing research risks. Adv Nurs Sci 2001:24:32–46.
Winickoff DE, Winickoff RN . The charitable trust as a model for genomic biobanks. N Engl J Med 2003;349:1180–1184.
Eiseman E, Bloom G, Brower J, Clancy N, Olmsted SS . RAND Science and Technology. Case Studies of Existing Human Tissue Repositories “Best Practices” for a Biospecimen Resource for the Genomic and Proteomic Era, 2003. http://biospecimens.cancer.gov/global/pdfs/RAND_REPORT.pdf. Accessed 2 September 2010.
Boggio A . Informing participants about research results. In: Elger B, Biller-Andorno N, Mauron A, Capron AM (eds). Ethical Issues in Governing Biobanks: Global Perspectives. Ashgate Publishing: Burlington, VT, 2008:189–196.
National Bioethics Advisory Commission. Research Involving Human Biological Materials: Ethical Issues and Policy Guidance, Vol. 1. Rockville, MD, 1999.
Clayton EW . Informed consent and biobanks. J Law Med Ethics 2005;33:15–21.
Organisation for Economic Co-Operation and Development. OECD Guidelines on Human Biobanks and Genetic Research Databases, 2009. http://www.oecd.org/dataoecd/41/47/44054609.pdf. Accessed 6 December 2011.
Accelerated Cure Project. Materials Transfer Agreement. http://www.acceleratedcure.org/repository/downloads/ACP_MTA.pdf. Accessed 27 March 2011.
Framingham Heart Study. Data and Materials Distribution Agreement. http://www.framinghamheartstudy.org/research/forms/dnadatamaterialsdist_agree.pdf. Accessed 23 March 2011.
NIDDK Central Repositories. Sample and Data Use Agreement, 1 December 2009. https://www.niddkrepository.org/niddkdocs/resources/NIDDK_Sample_Data_Distribution_AgreementFINAL.pdf. Accessed 6 December 2011.
NIMH Center for Collaborative Genetic Studies. Distribution Agreement, 24 January 2011. https://www.nimhgenetics.org/documents/Distribution%20Agreement.pdf. Accessed 5 April 2011.
Hallmans G, Vaught JB . Best practices for establishing a biobank. In: Dillner J (ed). Methods in Biobanking. Humana Press: Totowa, NJ, 2011: 241–260.
NHGRI Points to Consider for IRBs and Institutions in Their Review of Data Submission Plans for Institutional Certifications under NIH’s Policy for Sharing of Data Obtained in NHGRI-supported or Conducted Medical Sequencing Studies (NHGRI MSP), 29 November 2007. http://www.genome.gov/Pages/Research/SequenceMapsBAC/MedicalSequencing/MSPPtstoConsider03.12.08.pdf. Accessed 18 January 2012.
US Department of Health and Human Services. NIH policy for sharing of data obtained in NIH supported or conducted genome-wide association studies (GWAS). Federal Register 2007;72:49290–49097. http://grants.nih.gov/grants/guide/notice-files/NOT-OD-07-088.html. Accessed 6 December 2011.
Richardson HS, Cho MK . Secondary researchers’ duties to return incidental findings and individual research results: a partial-entrustment account. Genet Med, 2012;14:467–472.
Fabsitz RR, McGuire A, Sharp RR,et al. Ethical and practical guidelines for reporting genetic research results to study participants: updated guidelines from an NHLBI working group. Circ Cardiovasc Genet 2010;3: 574–580.
Yassin R, Lockhart N, González del Riego M,et al. Custodianship as an ethical framework for biospecimen-based research. Cancer Epidemiol Biomarkers Prev 2010;19:1012–1015.
Kohane IS, Mays DR, Altman RB . The incidentalome: a threat to genomic medicine. JAMA 2006;296:212–215.
Kohane IS, Taylor PL . Multidimensional results reporting to participants in genomic studies: getting it right. Science Translat Med 2010;2:1–4.
Caulfield T, McGuire AL, Cho M . Research ethics recommendations for whole-genome research: consensus statement. PLoS Biol 2008;6: 430–435.
Centers for Disease Control and Prevention. Genomic Testing: ACCE Model Process for Evaluating Genetic Tests, 2010. http://www.cdc.gov/genomics/gtesting/ACCE/index.htm. Accessed 10 December 2011.
NHRPAC Working Group on Genetics. IRB Guidebook Chapter on Human Genetics Research, Draft 2; 27 June 2002. http://www.hhs.gov/ohrp/archive/nhrpac/documents/nhrpac13.pdf. Accessed 17 January 2012.
Clinical Laboratory Improvement Amendments of 1988. Pub. L. No. 100-578, 102 Stat. 2903 (codified as amended at 42 U.S.C. § 263a (2006)).
US Department of Health and Human Services. Centers for Medicare and Medicaid Services. Conditions of participation for hospitals. 42 C.F.R. §§ 482 et seq.
Das S, Bale SJ, Ledbetter DH . Molecular genetic testing for ultra rare diseases: models for translation from the research laboratory to the CLIA-certified diagnostic laboratory. Genet Med 2008;10:332–336.
Rothstein MA . Tiered disclosure options to promote the autonomy and well-being of research subjects. Am J Bioethics 2006;6(6):20–21.
Clayton EW . Incidental findings in genetics research using archived DNA. J Law Med Ethics 2008;36:286–291.
US Department of Energy. Human Subjects Protection Resource Book, 3 May 2007. http://humansubjects.energy.gov/doe-resources/files/HumSubjProtect-ResourceBook.pdf. Accessed 6 December 2011.
Berg JS, Khoury MJ, Evans JP . Deploying whole genome sequencing in clinical practice and public health: meeting the challenge one bin at a time. Genet Med 2011;13:499–504.
Cassa CA, Savage SK, Taylor PL,et al. Disclosing pathogenic genetic variants to research participants: quantifying an emerging ethical responsibility. Genom Res 2012; e-pub ahead of print 6 January 2012
GENEVA Study. GENEVA Incidental Findings Files, 2009. https://genevastudy.org/Incidental_Findings_Files. Accessed 17 January 2012.
Knoppers BM, Kharaboyan L . Deconstructing’ biobank communication of results. SCRIPTed 2009;6:677–684.
Molnar MJ, Bencsik P . Establishing a neurological-psychiatric biobank: banking, informatics, ethics. Cell Immunol 2006;244:101–104.
Saykin AJ, Shen L, Foroud TM,et al. Alzheimer’s Disease Neuroimaging Initiative biomarkers as quantitative phenotypes: genetics core aims, progress, and plans. Alzheimers Dement 2010;6:265–273.
GENEVA Study Overview. https://www.genevastudy.org/StudyOverview. Accessed 17 January 2012.
Stack CB, Gharani N, Gordon ES, Schmidlen T, Christman MF, Keller MA . Genetic risk estimation in the Coriell Personalized Medicine Collaborative. Genet Med 2011;13:131–139.
Fullerton SM, Wolf WA, Brothers KB,et al. Return of individual research results from genome-wide association studies: experience of the Electronic Medical Records & Genomics (eMERGE) Network. Genet Med, this issue.
GENEVA Study. GENEVA Statement on Incidental Findings. https://genevastudy.org/Incidental_Findings_Files. Accessed 17 January 2012.
Mayo Clinic Biobank. Your Questions Answered. http://mayoresearch.mayo.edu/mayo/research/biobank/questions-answered.cfm. Accessed 5 April 2011.
Framingham Heart Study. Responsibilities of Ancillary Study Investigators, 2011. http://www.framinghamheartstudy.org/research/respons.html. Accessed 11 January 2012.
Thakuria J . Managing IFs and IRRs in genomic biobanks and archives. Presentation at working group meeting, Managing Incidental Findings and Research Results in Genomic Biobanks & Archives. Washington, DC, June 2010.
Wolf W . NUgene and the eMERGE Network–addressing incidental findings.Presentation at working group meeting, Managing Incidental Findings and Research Results in Genomic Biobanks & Archives. Washington, DC, June 2010.
Merz JF, Sankar P, Taube SE, Livolsi V . Use of human tissues in research: clarifying clinician and researcher roles and information flows. J Investig Med 1997;45:252–257.
Roberts JS, Shalowitz DI, Christensen KD,et al. Returning individual research results: development of a cancer genetics education and risk communication protocol. J Empir Res Hum Res Ethics 2010;5:17–30.
Cupples LA, Farrer LA, Sadovnick AD, Relkin N, Whitehouse P, Green RC . Estimating risk curves for first-degree relatives of patients with Alzheimer’s disease: the REVEAL study. Genet Med 2004;6:192–196.
Biesecker LG, Mullikin JC, Facio FM,et al. The ClinSeq Project: piloting large-scale genome sequencing for research in genomic medicine. Genome Res 2009;19:1665–1674.
Zawati MH, Rioux A . Biobanks and the return of research results: out with the old and in with the new? J Law Med Ethics 2011;39:614–620.
World Health Organization, International Agency for Research on Cancer. Common Minimum Technical Standards and Protocols for Biological Resource Centres Dedicated to Cancer Research, 2007. http://www.iarc.fr/en/publications/pdfs-online/wrk/wrk2/Standards_ProtocolsBRC.pdf. Accessed 16 March 2011.
The National Institute for Neurological Disorders and Stroke (NINDS) Human Genetics Initiative: Cell Line and DNA Repository. Frequently Asked Questions, 31 August 2011. http://ccr.coriell.org/Sections/Collections/NINDS/InvestigatorFAQ.aspx?PgId=150. Accessed 22 February 2012.
NUgene. How to Participate: FAQ. https://www.nugene.org/ppfaq.htm. Accessed 5 April 2011.
Vanderbilt University Medical Center. Vanderbilt BioVU. http://www.vanderbilthealth.com/main/25443. Accessed 23 March 2011.
Rhode Island Biobank. Frequently Asked Questions. http://ribb.brown.edu/Faqs.aspx. Accessed 10 January 2012.
Generation Scotland. Being a Volunteer. http://www.generationscotland.org/index.php?option=com_content&view=article&id=43&Itemid=50#BAP5. Accessed 10 January 2012.
O’Donnell C . Should we return individual research results and incidental findings from genomic biobanks and archives? Conference on Managing Incidental Findings and Research Results in Genomic Biobanks & Archives, Bethesda, MD, 19 May 2011. http://media2.law.umn.edu/jointdgr/mediaplayer/2011ifbb/player.php?src=05_09-ChristopherODonnell.mp4. Accessed 10 January 2012.
Marshfield Clinic. Guidance on Returning Research Results to Subjects, 2010. http://www.marshfieldclinic.org/rc/pages/default.aspx?page=policy_details.0&ID=3857. Accessed 10 January 2012.
National Institute of Environmental Health Sciences. Environmental Polymorphism Registry: Frequently Asked Questions. http://www.niehs.nih.gov/research/clinical/join/epr/participants/faq.cfm. Accessed 10 January 2012.
Beskow LM, Burke W, Merz JF,et al. Informed consent for population-based research involving genetics. JAMA 2001;286:2315–2321.
Acknowledgements
Preparation of this article was supported by the National Institutes of Health (NIH), National Human Genome Research Institute (NHGRI) grant 2-R01-HG003178 on “Managing Incidental Findings and Research Results in Genomic Biobanks & Archives” (S.M.W., Principal Investigator; J.P.K., F.L., B.V.N., Co-Investigators).
Thanks for invaluable input to all who presented at and participated in the project’s May 2011 conference in Bethesda, MD, on “Should We Return Individual Research Results and Incidental Findings from Genomic Biobanks & Archives?”; presenters are listed at http://www.lifesci.consortium.umn.edu/conferences/2011_ifbb/agenda. The complete list of the project members and presenters over the course of this project, listed with appreciation, is available at http://lifesci.consortium.umn.edu/ifbb-public. We are grateful for research assistance and support at various points in this project to Heather Bemmels, Rebecca Boxhorn, Leili Fatehi, Brian Grady, Cortney Jones, Christian Krautkramer, Gina Johnson, Pari McGarraugh, Michael Schubert, and Mao Thao. We are also indebted to Audrey Boyle for outstanding grants management.
Authors' institutions are listed on the first page of this article for identification only. The contents of this article are solely the responsibility of the authors and do not necessarily represent the views of NIH or NHGRI.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wolf, S., Crock, B., Van Ness, B. et al. Managing incidental findings and research results in genomic research involving biobanks and archived data sets. Genet Med 14, 361–384 (2012). https://doi.org/10.1038/gim.2012.23
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2012.23
Keywords
This article is cited by
-
Old and new challenges regarding comparable and viable data sharing in population-scale genomic research
European Journal of Human Genetics (2023)
-
Immortal data: a qualitative exploration of patients’ understandings of genomic data
European Journal of Human Genetics (2023)
-
Biobanking and risk assessment: a comprehensive typology of risks for an adaptive risk governance
Life Sciences, Society and Policy (2021)
-
The impact of reporting magnetic resonance imaging incidental findings in the Canadian alliance for healthy hearts and minds cohort
BMC Medical Ethics (2021)
-
Do patients and research subjects have a right to receive their genomic raw data? An ethical and legal analysis
BMC Medical Ethics (2020)
