
Abstract
Purpose: Fragile X syndrome is caused by expansion and subsequent methylation of a CGG trinucleotide repeat in the FMR1 5′-untranslated region. Southern blot analysis is typically required to determine expansion size for triplet repeat lengths >200. We describe a triplet-primed polymerase chain reaction-based method using automated capillary electrophoresis detection for qualitative assessment of expanded CGG repeats.
Methods: The assay uses triplet-primed polymerase chain reaction in combination with GC-melting reagents and substitution of 7-deaza-2-deoxyGTP for dGTP. Amplicons are resolved by capillary electrophoresis.
Results: A distinctive pattern of tapering or “stutter” polymerase chain reaction amplification was evident on capillary electrophoresis in male and female patients harboring all expanded allele lengths examined (up to 2000 CGG repeats) and could be used to differentiate normal, intermediate, premutation, and full mutation alleles. Full mutation alleles exhibited an additional late-migrating amplicon on capillary electrophoresis. Mixing experiments demonstrated sensitivity as low as 1% for detection of the full mutation allele. In a 1275-sample concordance study against our existing polymerase chain reaction platform (with Southern blot analysis for repeat lengths ≥55), the triplet-primed polymerase chain reaction method exhibited 100% concordance for normal, intermediate, expanded, and full mutation alleles. This method also detected the full mutation alleles in DNA isolated from blood spots.
Conclusion: This assay provides an accurate assessment of FMR1 repeat status and holds promise for use in carrier and newborn screening. The method distinguishes normal homozygous females from full mutation carrying females. Although the method is not useful for accurate sizing, it supplements the classic polymerase chain reaction method and results in significant reduction in the number of Southern blot analyses required to be performed in the laboratory to accurately assess the FMR1 genotype in all individuals.
Similar content being viewed by others


Main
Fragile X syndrome (FXS, OMIM 300624) is the most common inherited mental retardation syndrome, affecting ∼1:4000 men and ∼1:8000 women.1 The disease is caused by the expansion of a trinucleotide CGG repeat in the 5′-untranslated region of the FMR1 gene. Methylation of the expanded CGG tract leads to silencing of expression of the FMR1 gene. The American College of Medical Genetics2,3 defines a normal repeat length as between 5 and 44. Intermediate alleles of between 45 and 54 repeats almost never expand to full mutations in a single meiosis. Premutation alleles are defined as 55-200 CGG. Premutation alleles of <90 repeats have a variable risk for expansion to full mutations, whereas larger premutation alleles almost always expand when inherited through a female. Full mutation alleles are defined as >200 repeats. FXS is an X-linked dominant disease. Symptoms are usually milder in affected females and may consist only of attention deficit disorder or personality disorder. Men with FXS have profoundly delayed speech, mild to moderate mental retardation, and distinctive physical and behavioral traits. FXS is diagnosed more frequently in males than females. Thirty to 40% of male carriers of a premutation allele will suffer from fragile X-associated tremor and ataxia (FXTAS) by age 50 years,4,5 whereas 20% percent of female premutation carriers are affected with premature ovarian insufficiency (POI).6
Several studies have examined the carrier frequency of expanded alleles (≥55 repeats) in various populations. Observed carrier frequencies for FXS vary by population with 1:113 in Israel,7 1:259 in Quebec, Canada,8 and 1:317–1:382 in the United States.9,10 Because FXS is an X-linked disorder, every woman found to be a carrier is at risk for having affected offspring regardless of the genotype of her husband. In contrast, for recessive diseases, only couples in which both parents are carriers are at risk. Therefore, population-based carrier screening for FXS would yield a greater number of at-risk couples than cystic fibrosis in Caucasians (1:784 couples), Tay Sachs Disease in Ashkenazi Jewish couples (1:900 couples), or in diseases with the highest incidences where both the American College of Medical Genetics and American College of Obstetrics and Gynecology have recommended population-based carrier screening. Taking the lowest carrier frequency for FXS or 1:382 in the United States would yield approximately twice the number of at-risk Caucasian couples than cystic fibrosis and thrice the number of at-risk Ashkenazi Jewish couples for Tay Sachs disease. Interest in population-based carrier screening for FXS was shown and debated,11–13 and efforts to educate the public about FXS were conducted.14 We have experienced an increased demand for population-based carrier detection for FXS.15 Surveys have established that there is interest in prenatal screening and newborn screening for FXS.16–18 Despite educational campaigns, FXS remains difficult to diagnose for the majority of pediatricians. Kemper and Bailey18 demonstrated that only half of the doctors surveyed were aware that FXS could affect females and that only 28% of the surveyed doctors knew that premutation carriers might have symptoms of POI or FXTAS.
A recent study19 has shown that the age of diagnosis remains >3 years of age. Often, another sibling has been born by the time a proband with FXS is diagnosed. Newborn screening could lead to diagnosis in the newborn period and allow early intervention and family planning for future pregnancies. Population-based prenatal carrier detection could be cost-efficient20 and avoid the birth of affected children. Until recently, however, the technical difficulties of performing newborn screening20 and population-based carrier detection precluded offering them to the general population. Large expansions are refractory to polymerase chain reaction (PCR) amplifications. Thus, strategies for newborn screening were limited to screening only males and suffered from high false-positive rates,21–23 whereas strategies for population-based carrier screening for women required Southern blot analysis in addition to the PCR for each individual.
Recent advancements have overcome the technical limitations of FXS analysis. Recent studies have described new methods or improved on existing methods for the qualitative detection of expanded CGG repeats.21–24 We devised a method of capillary Southern analysis25 that was capable of identifying all female carriers of FXS. However, this technique was not suitable for newborn screening.
Several investigators have published PCR-based methods for detecting full CGG expansions. The first PCR assay to successfully amplify the full CGG expansion was described by Fu et al.26 The method used 7-deaza-2-deoxyGTP in addition to dATP, dCTP, dTTP, and dGTP and amplicons internally labeled with 32P-dCTP. This approach could detect up to 200 CGG repeats. Since then, attempts by various laboratories to improve on and automate this original method to detect larger CGG expansions or detect methylation status of CGG repeats was developed. Pergolizzi and coworkers27–31 used deaza-dGTP in PCR in place of dGTP, however, because amplicons with deaza-dGTP cannot be stained by ethidium bromide, this method requires Southern blot analysis of amplicons, thus limiting its utility. The use of triplet-primed PCR (TP-PCR) for the detection of CGG expansion in FMR1 was initially described by Daniels et al.,32 who coined the term “repeat primer PCR.” This technique involved nested PCR, using a reverse PCR primer having seven CCG repeats mixed with the forward primer of Fu et al.,26 to identify full mutations from as little as 20–40 buccal cells. The method showed 84% sensitivity of detection of full mutations.32 Other approaches for the detection of expansions, methylation status, or expression of FMR1 gene included the use of antibodies against FMR1 protein,33 interrogation of upstream regions to interpret methylation status of the CGG repeats in the FMR1 gene,34,35 methylation-specific PCR,36 or methylation-specific PCR incorporating TP-PCR for identification of methylated CGG alleles in the FMR1 gene.37,38 Recently, a modified Southern analysis method was used for rapid qualitative population-based screening of FMR1 gene.25 Two recent enhancements to the classic PCR methods were developed as shown using a combination of betaine and TP-PCR.21,22
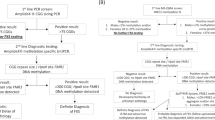
By using the wealth of knowledge in the literature, we were able to design a simple, rapid PCR-based method with capillary electrophoresis for the qualitative detection of expanded CGG repeats in the FMR1 gene. The method we describe is a single-tube assay that detects expansions and full mutations in both females and males, including mosaics. The method complements the classic PCR method by reducing the number of Southern blot analyses required to be performed as the method can be used to distinguish normal homozygous females from full mutation carrier females (as shown in Fig. 1). The method is robust, accurate, and appropriate for both prenatal population-based carrier screening and newborn screening programs.

Testing algorithm for samples submitted for fragile X testing. Samples received will be tested using two PCR reactions. Reaction 1 (in black) is for sizing normal, intermediate and small permutations and for gender determination,25 whereas Reaction 2 (CGG-PCR in blue) will detect the presence of expanded alleles and full mutations, without sizing. Samples showing evidence of stutter or full mutation amplification will be reflexed to Southern blot analysis for methylation status and sizing. As shown in the figure, the incorporation of the triplet-primed PCR method described here significantly reduces the number of Southern blot analyses performed in the laboratory as the method clearly distinguishes normal homozygous females, which in our experience represent ∼99% of all female samples,25 from full mutation carriers.
MATERIALS AND METHODS
Clinical samples
Residual archived DNA from patient samples already processed for fragile X analysis using our existing assays were de-identified and used in the development of the new method. DNA was extracted using the Qiagen M96 robot and reagents, Qiagen Gentra robot and reagents, and Qiagen 9604 reagents (Qiagen, Carlsbad, CA).
Assay conditions
The PCR step of this assay uses the 7-deaza-2-deoxy guanosine triphosphate (deaza-dGTP) in place of dGTP, a combination of Q-solution and dimethyl sulfoxide (DMSO) in the PCR mix, and an optimized cycling program. The Roche FastStarTaq kit (Roche Applied Science, Indianapolis, IN) reagents were used in the PCR mix. The PCR mix contained 1× PCR buffer; 2 mM MgCl2; 6% DMSO (Sigma); 1.7% Q solution (Qiagen); 0.2 mM each of deaza-dGTP, dATP, dCTP, and dTTP; 1 unit of FST polymerase; and 0.6 μM each of the forward primer (FMR1F), reverse primer 1 (FMR1R) or 2 (FMR1CCGR), and M13 reverse linker primer:
-
FMR1F: TGTAAAACGACGGCCAGT GCTCAGCTCCGTTTCGGTTTCACTTCCGGT;
-
FMR1R: CAGGAAACAGCTATGACC CTCGAGGCCCAGCCGCCGCCGCC;
-
FMR1CCGR: CAGGAAACAGCTATGACC CCGCCGCCGCC; and
-
M13 reverse linker primer: CAGGAAACAGCTATGACC.
The underlined sequence of FMR1F is the C primer of Fu et al.26; the underlined sequence of FMR1R is from bases 13882–13904 of GenBank accession number L29074.1, corresponding to the junction sequence of the 3′-end of the CGG repeat tract and flanking sequence; and the underlined sequence of FMR1CCGR represents almost 4 units of reverse CCG repeats that hybridize randomly across the CGG repeat tract (TP-PCR). The M13 reverse linker primer corresponds to the linker sequence of reverse primers and helps to boost the signals of the fragments amplified. Three microliters of genomic DNA (10–150 ng totally) was added for a final PCR reaction volume of 15 μL.
The cycling program, performed on an ABI 9700 thermal cycler, was started by incubating the mix at 98°C for 10 minutes to activate the polymerase, followed by 10 cycles of denaturation at 97°C for 35 seconds, annealing at 64°C for 2 minutes, and extension at 68°C for 8 minutes, followed by 25 cycles of denaturation at 97°C for 35 seconds, annealing at 64°C for 2 minutes, and extension at 68°C for 8 minutes 20 seconds (with 20 seconds extension each additional cycle). For all 35 cycles, the ramp rate was adjusted to 64% for the denaturation and extension steps (ramping up at 0.5°/second), and to 25% for the annealing step (ramping down at 0.5°C/second).
Two microliters of the PCR product (sometimes diluted 1:5 or 1:10 in H2O) were mixed with HiDi Formamide and Rox 1000 size standard (Celera), and samples were injected on an ABI 3730 DNA Analyzer equipped with a 36-cm capillary loaded with Pop-7 polymer (Applied Biosystems) at 3 V for 7 seconds. Data were analyzed using GeneMapper software (Applied Biosystems). The Rox 1000 size standard, although not used for accurate sizing, is helpful in locating full mutation peaks, which are usually located around 1050 bases on the electropherogram. Rox 600 (Applied Biosystems) or MapMarker1000 (Bioventures, Inc., Murfreesboro, TN) can also be used. Although full mutations ≥200 repeats will have a composition of at least 770 bp using our primers, all full mutation alleles examined appear at around 1050 bases. That is because of the limitation of the capillary. We used a 50-cm capillary as well to determine whether we can size better the full mutation peaks, but all full mutation peaks appeared around the 1050 bases.
A panel of anonymized, previously genotyped samples were used for initial development of the PCR method. The panel included a homozygous normal female (28/28 repeats), a heterozygous normal female (18/21), a premutation female (31/64), full mutation females (19/450 and 29/530), a normal male (28), a premutation male (74), and mosaic full mutation male (105/350). Once conditions for the PCR were established, a larger set of 47 anonymized, previously genotyped samples (Table 1), weighted toward full mutation carriers (male or female) was used to test the robustness of the assay.
To evaluate assay sensitivity to mosaicism, we made three DNA sets using archived, previously genotyped samples, each set harboring two DNA samples mixed at different ratios. The first set contained DNA samples from two males, one from a normal male with 28 CGG repeats, and one from affected male with 850 CGG repeats; set B contained two males, a normal male (28 CGG) and affected male (853); and set C contained two female samples, a normal (28/28) and an affected female (19/450).
Detection from blood spots
Whole blood from previously genotyped, residual samples was spotted on blood cards. Blood spots were generated using the BSD1000 GenePunch instrument (BSD Robotics, Australia). DNA was extracted using BioSprint 96 DNA Blood Kit reagents from Qiagen.
RESULTS
Assay development
The concept of the new method is similar to the TP-PCR methods initially described for fragile X32 and then applied for other triplet repeat-containing genes.39,40 Figure 2 shows the location of FMR1F and FMR1R primers and the concept of the method. Two reverse primers were tested, each harboring almost four CCG-units; the difference between FMR1R and FMR1CCGR is that the FMR1R primer included FMR1 sequences downstream of the CGG repeat tract, with the concept that presence of both triplet CCG sequences and downstream sequences might facilitate unwinding of the CGG secondary structure at the junction point and could also provide better specificity for FMR1 sequences. The FMR1CCGR primer contains only triplet CGG repeats in addition to the linker. Daniels et al.32 used a 7-CCG repeat primer, whereas our triplet primer contains almost four CCG units. FMR1R contains both CGG triplets in addition to FMR1 gene-specific nonrepeat sequences. Once the initial round of PCR is complete, longer templates are generated with more CCG units that can serve themselves as primers and templates for further cycling rounds (Fig. 2).

Schematic representation of the triplet-primed PCR method. Forward and reverse primers are shown. The reverse primer hybridizes at the junction of 3′-end of CGG tract and down stream sequences, but it can also hybridize randomly across the CGG tract. After initial rounds of PCR, extended reverse primers can themselves serve as primers. This results in various PCR product sizes giving the “stutter” pattern on electrophoresis on the ABI 3730.
We initially attempted to devise a PCR mix for amplification of all normal, premutation, and full mutations of the CGG tract by formulating various combinations of mixes using Roche Expand polymerase PCR mix buffer 2 or 3 and Roche High-GC PCR mix, and incorporating various additives at different concentrations including Roche Q-solution, DMSO, HiDi formamide, Tween-20, or combinations thereof. We first settled on the use of Roche Expand kit without the addition of deaza-dGTP. We then tested the use of the Roche FastStarTaq kit reagents and found better performance in terms of signal intensity under identical cycling conditions (data not shown). The inclusion of the M13 linker in the reverse primers also helped boost the signal by approximately 30%. The incorporation of ramping in the cycling program, similar to the process we described previously in a different assay,41 helped further boost signal amplification. Ramping is a PCR cycling parameter that allows the user to control the rate at which the thermocycler moves from one temperature to another. The default ramping parameter is as fast as the machine is capable of changing temperature. Slowing the ramping rate gives more time for primers to bind and rebind to their substrate. In this particular assay, controlling the ramping rate is critical to the robustness of the assay.
Other primers tested include primers containing 7-CCG units, 10-CCG units, and others. All primers showed stutter amplification indicative of presence of premutation or full mutation. All the above assay-optimization experiments and data are not shown but are available on request.
Development of a new triplet-primed PCR for qualitative assessment of FMR1 CGG expansion status
After identifying standard conditions for the PCR mix, we were able detect a stutter pattern of amplification whenever a premutation or a full mutation was present in a DNA sample. However, because of the complexity of amplifying the CGG tract, we decided to test using deaza-dGTP in the PCR reaction in place of dGTP. Figure 3 shows the effect of addition of deaza-dGTP to the reaction mix of a heterozygote female carrier (29/530). Increasing the concentration of deaza-dGTP not only boosted the signal from stutter amplification but also led to the appearance of a peak that migrated very late on the ABI 3730 capillary and was evident only when a full mutation was present (Figs. 3 and 4). Because the capillary instrument does not rely on ethidium bromide staining to visualize DNA, we are able to detect the presence of a premutation and a full mutation in single PCR reaction containing deaza-dGTP by scrutinizing stutter and full mutation signals. It should be noted that the method does not size alleles correctly. The method, being qualitative, is used as screening method. By using this method, we were able to distinguish homozygous normal females from heterozygous full mutation carrier females in an assay sufficiently robust for population-based carrier screening.

Effect of deaza-dGTP on full mutation detection. PCR master mixes were formulated with shown concentrations of 7-deaza-dGTP and dGTP. An affected female DNA sample (19/450) was used in the PCR reactions. Normal and intermediate alleles migrate in the gray-highlighted zone of the electropherogram (<55 CGG repeats), whereas premutation and full mutation alleles are present in the pink-highlighted area of the electropherogram (≥55 CGG repeats). As shown, tapering stutter amplification and full mutation signal intensities increase with increasing deaza-dGTP concentration. All electropherograms are zoomed to 500 relative fluorescence units (RFU) to show stutter and full mutation amplification. Full range signal intensity (>20,000 RFU) figures are available on request. The arrow shows the signal for full mutation. Replacing dGTP with deaza-dGTP influences the migration of the amplified full mutation fragment.

Example of results obtained from normal, carrier, and affected female and male patients. The status of alleles in each patient is shown. The assay distinguishes between normal females or males, including normal homozygous females, from full mutation carrier females or males. All electropherograms are zoomed to 1,000 RFU to show stutter and full mutation amplification. Full range signal intensity (>20,000 RFU) figures are available on request. The last panel shows a sample from a mosaic male with 105/350 alleles. The 105-repeat allele is marked by an asterisk.
We tested various sizes of CGG triplet repeats by using a panel of eight anonymized, previously genotyped samples. In Figure 4, the gray-shaded area represents the range of normal and intermediate allele CGG repeats numbers (≤54), and the pink-shaded area represents expanded CGG repeats (either premutation or full mutation alleles). As shown in Figure 4, normal and intermediate alleles are present only in gray part of the figure, whereas premutation and full mutation alleles (with ≥55 CGG repeats) are present in the pink area of the electropherogram that denotes expanded alleles. Normal females and normal males demonstrate alleles only in the gray area of the electropherogram, whereas premutation males and females have alleles in the pink area, with stutter amplification before the actual premutation peak. Full mutation carrying males and females display, in addition to normal allele in females, a tapering stutter amplification followed by a signal at the end of electropherogram, which is present only when a full mutation is present. Most significantly, normal homozygote females can be distinguished from full mutation carrier females using this method (Fig. 4).
To establish assay robustness for the detection of the CGG status, we tested the assay on previously genotyped DNA samples. The individual performing the test was blinded to the status of CGG repeats and the results scored by a second individual also blinded to the previously determined genotype. The panel of 47 anonymized samples contained homozygous females (N = 3), normal heterozygous females (N = 1), intermediate female (N = 1), premutation females (N = 4), and full mutation females (N = 11), in addition to intermediate male (N = 1), premutation male (N = 2), and full mutation males (N = 24; Table 1). All samples were identified correctly, demonstrating the capability of the new method in identifying presence of premutations and full mutations in all conditions.
Detection of mosaics and assay sensitivity
One concern for FXS analysis is the ability to detect mosaic alleles of permutation and full mutations that are sometimes identified using sensitive Southern blot analysis. Figure 4 shows the results of a mosaic male with a premutation allele of 105 repeats and a full mutation allele of 350, identified from a blood sample. Table 1 and Figure 5 also show samples with mosaic full mutations including two male samples (samples 15 and 17) and two female samples (samples 2 and 7). Sample 17, with mosaic for 78 and 2000 alleles, showed amplification signal for the longest full mutation, which we have in our archived collection. The signal for the full mutation was weak but detectable. Previous Southern blot analysis of this sample showed the unmethylated premutation allele accounted for 60% of alleles, whereas the methylated full mutation 2000 repeat allele accounted for the rest in this affected male sample. The method also detected mosaic premutations in full mutation carrier females (Fig. 5). These results show that the new PCR method is able to detect mosaic alleles in both males and females.

Detection of mosaic males and females. Samples 15 and 17 from male patients and samples 2 and 7 from female patients (Table 2) with mosaic premutation and full mutations are shown. Sample 17 shows weak signal for full mutation. It is not clear whether the DNA quality or the level of mosaicism of this archived sample affected robustness of amplification of the full mutation, but the signal for full mutation is present, albeit weakly. Other samples show robust stutter and full mutation amplification. Premutations are marked by an asterisk, whereas mutations are marked by an arrow.
We also tested the sensitivity of this assay to low level mosaicism by performing artificial mixing experiments using DNA samples from males and females with normal and full mutations, thereby creating artificial mosaics. We constructed three DNA sets using archived, previously genotyped samples, each set harboring two DNA samples mixed at different ratios; set “A” contained DNA samples from two males, one from a normal male with 28 CGG repeats and one from affected male with 850 CGG repeats; set “B” contained two different male DNAs, a normal male (28 CGG repeats) and affected male (853 CGG repeats), and set “C” contained two female samples, a normal (28/28 repeats) and an affected female (19/450 repeats).
Figure 6 demonstrates the results from these samples artificial mosaic series. For male DNA, the full mutation was detectable at levels of 5% in set “A” and 10% in set “B.” As shown in Figure 6, the intensity of the stutter amplification and the full mutation signals increase as the fraction of full mutation DNA increases. In set “C,” from mixed female samples, the assay is capable of detecting as low as 1% mosaic full mutation content in female DNA that contained alleles of three lengths (19, 28, and 450 repeats), where the smaller, normal alleles would have been favored during PCR. At 1% mosaic content, a weak signal from a full mutation is present (Fig. 6C), whereas stutter amplification is appearing reliably at 10% content of the full mutation. On average, we were able to detect full mutation expansion, by both the stutter amplification and full mutation signal detection, at levels as low as 5–10% in both males and females.

Assay sensitivity to low levels of full mutations. DNA samples from normal males or females were mixed with affected males or females to give final concentration of full mutation content as shown in each panel. The gray-highlighted region of the electropherograms shows full scale RFU to show normal alleles, whereas the mutant, pink area is shown at 1000 RFU to show mutant and stutter amplification. A, Set A from mixed normal male (28 repeats) and affected male (850 repeats) shows detection of full mutation at 5% “mosaic” content. B, Set B from mixed normal male (28 repeats) and affected male (853 repeats) shows “mosaic” detection from males at 10%. Note the disappearance of the normal 28-repeat allele (black arrow) as the percentage of full mutation is increased. C) Set C from mixed normal homozygous female (28/28 repeats) and affected female (19/450 repeats) shows detection of full mutation in female DNA at 1% “mosaic” content, although the full mutation allele represents only one of the four alleles present in the mix and at very low amounts in the low “mosaic content. The presence of the 19-repeat allele (black arrow) and 28-repeat allele (red arrow) is shown.
Concordance study
In developing any new platform for assays with anticipated high volume, our laboratory performs a 1000-sample concordance study to test and compare assay performance with existing platforms.25,42–44 We, therefore, evaluated the new method by performing a platform comparison of 1275 consecutive, anonymized whole blood samples submitted for fragile X analysis using our current platform composed of a two-step process: a PCR incorporating gender marker and a reflex to Southern blot analysis for any sample with an expansion length of 45 repeats or above and for any apparent homozygous females. There was 100% concordance with the existing method. As tabulated in Table 2, the series contained a total of six patients (three female and three male) harboring full mutations.
Detection from blood spots and saliva
Once we had technically validated the assay on peripheral blood, we attempted two alternate sample types, saliva and newborn blood spots. As we did not have access to prespotted Guthrie cards from FXS patients, we first spotted residual blood from previously genotyped patients on Guthrie cards. Blood spots were punched from each card using BSD1000 GenePunch instrument to generate two punches (3.2 mm each) per patient, and DNA was extracted using Qiagen BioSprint reagents. DNA from blood spots was analyzed by the new PCR method. A total of 37 samples containing the varying FMR1 CGG expansions in males and females were tested (Table 3). The series contained 14 samples with intermediate repeats, nine samples with premutations, and four samples with full mutations (as determined by Southern blot analysis), one female (20/500 repeats) and three males (300–700; 1250; 1250). Figure 7 shows an example of the results obtained. All the samples were genotyped by the new qualitative method correctly as harboring normal, intermediate, premutation, or full mutations.

Detection of normal, premutation, and full mutation carriers from blood spots. Blood samples from samples (given in Table 3) were spotted on filter cards and punched using BSD1000 GenePunch. DNA from two spots was extracted and PCR performed. Number in each panel corresponds to numbers in Table 3. Also shown is allele content in each sample. The new method can qualitatively identify full mutations and distinguish normal females, including homozygous females, from carrier females.
DNA prepared from saliva can be successfully amplified in the new assay, but we do not have any samples from affected individuals or carriers, hence, we cannot confirm the ability to detect full mutations and premutations in saliva reliably at this time. We have no reason to believe that there will be any problems however.
DISCUSSION
The method we describe here is a rapid, single-tube method capable of detecting the full spectrum of FMR1 CGG repeat instability, up to 2000 repeat units, which was the largest sample that we possessed in our archived DNA collection. The method also allowed detection of mosaic males and females from whole blood. The method was also capable of detecting premutations and full mutations from blood spots, which would facilitate newborn screening. The method is capable of rapid turn around time as it does not require Southern blot analysis. We performed an extensive technical validation of the method by testing >1300 samples, with various repeat sizes, and all samples with full mutation alleles, regardless of sex, were correctly identified. This is important, as the true technical impediment for implementing population-based carrier screening or newborn screening using prior methods has been distinguishing truly homozygous females from carrier females.
The method we describe is similar to the nested-repeat primer PCR method developed previously for attempting fragile X screening from single cells.32 In that report, the authors noted that, in samples with full mutations, “the fragile X DNA produce a more complex band pattern, with extra bands of both higher and lower molecular weight.”32 However, in that method, distinguishing full mutation allele from normal one was not very clear using agarose gels because of weaker signal and background fluorescence, which lead to assay sensitivity of 84% using DNA from 20–40 buccal epithelial cells.32 However, the authors of that report were attempting to design a method for analysis of expansions from single cells for preimplanation genetic diagnosis32 and did not report testing the method on whole bloods. In our method, we used an optimized PCR mix and cycling program without nested amplification. Analysis on capillary electrophoresis instrument using fluorescently labeled amplicons facilitated identifying stutter amplification and full mutation signals using a single PCR tube. We achieved 100% sensitivity, as every sample we tested with full mutation was identified, whether male or female.
Our testing paradigm calls for two PCR reactions per patient run simultaneously (Fig. 1). One reaction, described previously,25 will allow accurate sizing of repeat number in the normal, intermediate, and small premutation zone and, also, gender identification/confirmation. The second PCR reaction is the method described here for identification of presence of larger premutations and full mutations. The combination of two PCR reactions per patient yields results for both males and females, and this would serve the purpose for carrier and/or newborn screening.
For newborn samples showing evidence of full mutation, further follow-up will be recommended, as the amount of available DNA from blood spots will not be sufficient to perform Southern blot analysis. This is similar to the approach of cystic fibrosis newborn screening programs, where a positive immunoreactive trypsinogen test prompts secondary tests of sweat chloride and/or cystic fibrosis transmembrane conductance regulator mutation identification. Although a recent report35 described the use of methylation-specific PCR for detection of FMR1 promoter methylation and FXS incidence in newborns from the state of Georgia, the method, although promising, is laborious, even with pooling of samples, as it relies on the multistep bisulfite conversion of DNA, and the method can miss female samples carrying full mutations, because some full mutation carrying females displayed methylation levels within the range of normal females.35
The method described here is not intended as a stand alone test for diagnostic purposes. Rather, the method is used as a qualitative assessment for repeat status, which would prompt, on a positive finding of a premutation or full mutation, further follow-up of methylation status and accurate sizing. Although the sizing of full mutation has little or no effect on phenotype presentation,45 it is important to determine methylation status of the CGG repeats, and therefore Southern blot analysis will be performed. We are developing an alternative method for assessing methylation status without performing a Southern blot using methylation-sensitive restriction digestion followed by PCR using primers flanking the restriction site. Only methylated DNA yields PCR product under these conditions. Because the actual size of the expanded repeats has little clinical relevance,45 a combination of these techniques has the potential to eliminate the need for Southern blotting altogether in fragile X analysis.
Even without the new methylation assay, using the technique described in this article will drastically reduce the number of Southern blots necessary in a FXS screening program. In our laboratory, only approximately 2% of males and 0.5% of females have expanded alleles.14 Therefore, approximately 99% of Southern blots can be eliminated in a program screening an equal number of males and females.
We have described a method that overcomes the technical block to instituted programs of newborn screening or population-based carrier detection for FXS. Now the medical, ethical, and legal issues can be investigated and discussed as suggested by Bailey et al.46 The issues have not changed dramatically since the early 1990s, when carrier/prenatal screening was proposed and debated,11–13,47–49 in terms of the incomplete and variable presentation of the FXS phenotype in full mutation carrier females, and of the unpredictable presentation in mosaic males. Another issue is related to stigmatizing carriers of premutations, biases in interaction by peers, or overprotection by parents.46
Questions will arise such as would having such information, if newborn screening is implemented, be beneficial for early intervention, or would this cause certain perceived biases against such individuals? Would informing parents of children who happen to harbor the premutation alleles in the FMR1 benefit or harm the child?
Consenting to prenatal testing in the span of a routine office visit during pregnancy or to newborn screening at hospital admission for delivery will certainly not be sufficient to understand the full impact of such screening. It certainly will be a challenge to educate the public about the complexities of fragile X testing, as the issues involved are not only just related to mental retardation but also have neuropsychologic (memory and function), physical (FXTAS), and reproductive (POI) implications, in addition to involvement of FXS in autism, as was reviewed recently.46 Several recent studies have discussed prenatal and newborn screening issues and attitudes,17,50 and more studies will be needed if, and when, population and/or newborn screening for FXS is implemented. It is likely that a wide effort to educate the public, during pregnancy at the latest, will be needed to allow enough understanding of the issues, and it might be beneficial to allow voluntary participation in such programs initially until better understanding of implementing such programs is achieved.
In summary, we designed a simple, rapid, high throughput, single-tube assay for the qualitative detection of CGG repeat status in both males and females, suitable for prenatal carrier screening and for newborn screening programs. A recommendation from American College for Medical Genetics or American College of Obstetrics and Gynecology for implementing prenatal screening will likely await further studies of performance of such new tests.
References
Turner G, Webb T, Wake S, Robinson H . Prevalence of fragile X syndrome. Am J Med Genet 1996; 64: 196–197.
Maddalena A, Richards CS, McGinniss MJ, et al. Technical standards and guidelines for fragile X: the first of a series of disease-specific supplements to the Standards and Guidelines for Clinical Genetics Laboratories of the American College of Medical Genetics. Quality Assurance Subcommittee of the Laboratory Practice Committee. Genet Med 2001; 3: 200–205.
Sherman S, Pletcher BA, Driscoll DA . Fragile X syndrome: diagnostic and carrier testing. Genet Med 2005; 7: 584–587.
Hagerman PJ, Hagerman RJ . Fragile X-associated tremor/ataxia syndrome (FXTAS). Ment Retard Dev Disabil Res Rev 2004; 10: 25–30.
Hagerman RJ, Leavitt BR, Farzin F, et al. Fragile-X-associated tremor/ataxia syndrome (FXTAS) in females with the FMR1 premutation. Am J Hum Genet 2004; 74: 1051–1056.
Allingham-Hawkins DJ, Babul-Hirji R, Chitayat D, et al. Fragile X premutation is a significant risk factor for premature ovarian failure: the International Collaborative POF in Fragile X study—preliminary data. Am J Med Genet 1999; 83: 322–325.
Toledano-Alhadef H, Basel-Vanagaite L, Magal N, et al. Fragile-X carrier screening and the prevalence of premutation and full-mutation carriers in Israel. Am J Hum Genet 2001; 69: 351–360.
Rousseau F, Rouillard P, Morel ML, Khandjian EW, Morgan K . Prevalence of carriers of premutation-size alleles of the FMRI gene–and implications for the population genetics of the fragile X syndrome. Am J Hum Genet 1995; 57: 1006–1018.
Crawford DC, Meadows KL, Newman JL, et al. Prevalence and phenotype consequence of FRAXA and FRAXE alleles in a large, ethnically diverse, special education-needs population. Am J Hum Genet 1999; 64: 495–507.
Cronister A, DiMaio M, Mahoney MJ, Donnenfeld AE, Hallam S . Fragile X syndrome carrier screening in the prenatal genetic counseling setting. Genet Med 2005; 7: 246–250.
Turner G, Robinson H, Laing S, et al. Population screening for fragile X. Lancet 1992; 339: 1210–1213.
Howard-Peebles PN, Maddalena A, Black SH, Schulman JD . Population screening for fragile-X syndrome. Lancet 1993; 341: 770.
Spence WC, Black SH, Fallon L, et al. Molecular fragile X screening in normal populations. Am J Med Genet 1996; 64: 181–183.
Keenan J, Kastner T, Nathanson R, Richardson N, Hinton J, Cress DA . A statewide public and professional education program on fragile X syndrome. Ment Retard 1992; 30: 355–361.
Strom CM, Crossley B, Redman JB, et al. Molecular testing for fragile X syndrome: lessons learned from 119,232 tests performed in a clinical laboratory. Genet Med 2007; 9: 46–51.
Skinner D, Sparkman KL, Bailey DB Jr, Screening for fragile X syndrome: parent attitudes and perspectives. Genet Med 2003; 5: 378–384.
Fanos JH, Spangner KA, Musci TJ . Attitudes toward prenatal screening and testing for fragile X. Genet Med 2006; 8: 129–133.
Kemper AR, Bailey DB Jr, Pediatricians' knowledge of and attitudes toward fragile X syndrome screening. Acad Pediatr 2009; 9: 114–117.
Bailey DB Jr, Raspa M, Bishop E, Holiday D . No change in the age of diagnosis for fragile X syndrome: findings from a National Parent Survey. Pediatrics 2009; 124: 527–533.
Musci TJ, Caughey AB Cost-effectiveness analysis of prenatal population-based fragile X carrier screening. Am J Obstet Gynecol 2005; 192: 1905–1512; discussion 1912–1915.
Saluto A, Brussino A, Tassone F, et al. An enhanced polymerase chain reaction assay to detect pre- and full mutation alleles of the fragile X mental retardation 1 gene. J Mol Diagn 2005; 7: 605–612.
Tassone F, Pan R, Amiri K, et al. A rapid polymerase chain reaction-based screening method for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J Mol Diagn 2008; 10: 43–49.
Latham GJ, Filipovic-Sadic S, Sah S et al. Evaluation of a novel FMR1 PCR assay that can amplify Fragile X full mutation. Paper presented at: ACMG Annual Clinical Genetics Meeting. Tampa, FL, March 25–29, 2009.
Lyon E, Yu P, Jama M et al. A rapid PCR assay suitable for Fragile X population screening. Paper presented at: ACMG Annual Clinical Genetics Meeting. Tampa, FL, March 25–29, 2009.
Strom CM, Huang D, Li Y, et al. Development of a novel, accurate, automated, rapid, high-throughput technique suitable for population-based carrier screening for Fragile X syndrome. Genet Med 2007; 9: 199–207.
Fu YH, Kuhl DP, Pizzuti A, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell 1991; 67: 1047–1058.
Pergolizzi R, Brown WT, Goonewardena P, et al. Molecular characterization of a DNA probe, U6.2, located close to the fragile X locus. Am J Med Genet 1991; 38: 380–383.
Erster SH, Brown WT, Goonewardena P, Dobkin CS, Jenkins EC, Pergolizzi RG . Polymerase chain reaction analysis of fragile X mutations. Hum Genet 1992; 90: 55–61.
Pergolizzi RG, Erster SH, Goonewardena P, Brown WT . Detection of full fragile X mutation. Lancet 1992; 339: 271–272.
Snow K, Doud LK, Hagerman R, et al. Analysis of a CGG sequence at the FMR-1 locus in fragile X families and in the general population. Am J Hum Genet 1993; 53: 1217–1228.
Brown WT, Houck GE Jr, Jeziorowska A, Pergolizzi RG, Erster SH, Thibodeau SN . Rapid fragile X carrier screening and prenatal diagnosis using a nonradioactive PCR test. JAMA 1993; 270: 1569–1575.
Daniels R, Holding C, Kontogianni E, Monk M . Single-cell analysis of unstable genes. J Assist Reprod Genet 1996; 13: 163–169.
Jenkins EC, Wen GY, Kim KS, et al. Prenatal fragile X detection using cytoplasmic and nuclear-specific monoclonal antibodies. Am J Med Genet 1999; 83: 342–346.
Strelnikov V, Nemtsova M, Chesnokova G, Kuleshov N, Zaletayev D . A simple multiplex FRAXA, FRAXE, and FRAXF PCR assay convenient for wide screening programs. Hum Mutat 1999; 13: 166–169.
Coffee B, Keith K, Albizua I, et al. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am J Hum Genet 2009; 85: 503–514.
Das S, Kubota T, Song M, et al. Methylation analysis of the fragile X syndrome by PCR. Genet Test 1997; 1: 151–155.
Zhou Y, Lum JM, Yeo GH, et al. Simplified molecular diagnosis of fragile X syndrome by fluorescent methylation-specific PCR and GeneScan analysis. Clin Chem 2006; 52: 1492–1500.
Zhou Y, Law HY, Boehm CD, Kiing J, Tay SK, Chong SS . Robust fragile X (CGG)n genotype classification using a methylation specific triple PCR assay. J Med Genet 2004; 41: e45.
Ciotti P, Di Maria E, Bellone E, Ajmer F, Mandich P . Triplet repeat primed PCR (TP PCR) in molecular diagnostic testing for Friedreich ataxia. J Mol Diagn 2004; 6: 285–289.
Falk M, Vojtísková M, Lukás Z, Kroupová I, Froster U . Simple procedure for automatic detection of unstable alleles in the myotonic dystrophy and Huntington's disease loci. Genet Test 2006; 10: 85–97.
Hantash FM, Redman JB, Starn K, et al. Novel and recurrent rearrangements in the CFTR gene: clinical and laboratory implications for cystic fibrosis screening. Hum Genet 2006; 119: 126–136.
Strom CM, Clark DD, Hantash FM, et al. Direct visualization of cystic fibrosis transmembrane regulator mutations in the clinical laboratory setting. Clin Chem 2004; 50: 836–845.
Strom CM, Janeczko RA, Anderson B, et al. Technical validation of a multiplex platform to detect thirty mutations in eight genetic diseases prevalent in individuals of Ashkenazi Jewish descent. Genet Med 2005; 7: 633–639.
Strom CM, Janeszco R, Quan F, et al. Technical validation of a TM Biosciences Luminex-based multiplex assay for detecting the American College of Medical Genetics recommended cystic fibrosis mutation panel. J Mol Diagn 2006; 8: 371–375.
Fisch GS, Carpenter N, Howard-Peebles PN, et al. Lack of association between mutation size and cognitive/behavior deficits in fragile X males: a brief report. Am J Med Genet 1996; 64: 362–364.
Bailey DB Jr, Skinner D, Davis AM, Whitmarsh I, Powell C . Ethical, legal, and social concerns about expanded newborn screening: fragile X syndrome as a prototype for emerging issues. Pediatrics 2008; 121: e693–e704
Bonthron D, Strain L . Population screening for fragile-X syndrome. Lancet 1993; 341: 769.
Bundey S, Norman E . Population screening for fragile-X syndrome. Lancet 1993; 341: 770.
Palomaki GE, Haddow JE . Is it time for population-based prental screening for fragile-X?. Lancet 1993; 341: 373–374.
Anido A, Carlson LM, Sherman SL . Attitudes toward fragile X mutation carrier testing from women identified in a general population survey. J Genet Couns 2007; 16: 97–104.
Acknowledgements
We thank Jeff Radcliff (Quest Diagnostics Nichols Institute) for reviewing and editing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: Some of the authors on this manuscript hold stocks or stock options in Quest Diagnostics and all authors are employees of Quest Diagnostics Nichols Institute. C. M. Strom is an inventor on a patent application (US 20080124709) related to Southern capillary analysis for FXS. No other financial or conflict of interest issues exist.
Rights and permissions
About this article
Cite this article
Hantash, F., Goos, D., Tsao, D. et al. Qualitative assessment of FMR1 (CGG)n triplet repeat status in normal, intermediate, premutation, full mutation, and mosaic carriers in both sexes: Implications for fragile X syndrome carrier and newborn screening. Genet Med 12, 162–173 (2010). https://doi.org/10.1097/GIM.0b013e3181d0d40e
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e3181d0d40e
Keywords
This article is cited by
-
Genetic modifiers in rare disorders: the case of fragile X syndrome
European Journal of Human Genetics (2021)
-
Assessment of FMR1 triplet repeats in patients affected with mental retardation, fragile X syndrome and primary ovarian insufficiency
Journal of Genetics (2020)
-
Abnormally Methylated FMR1 in Absence of a Detectable Full Mutation in a U.S.A Patient Cohort Referred for Fragile X Testing
Scientific Reports (2019)
-
Short stature and primary ovarian insufficiency possibly due to chromosomal position effect in a balanced X;1 translocation
Molecular Cytogenetics (2015)
-
EMQN best practice guidelines for the molecular genetic testing and reporting of fragile X syndrome and other fragile X-associated disorders
European Journal of Human Genetics (2015)
