
Abstract
Dilated cardiomyopathy (DCM), usually diagnosed as idiopathic dilated cardiomyopathy (IDC), has been shown to have a familial basis in 20–35% of cases. Genetic studies in familial dilated cardiomyopathy (FDC) have shown dramatic locus heterogeneity with mutations identified in >30 mostly autosomal genes showing primarily dominant transmission. Most mutations are private missense, nonsense or short insertion/deletions. Marked allelic heterogeneity is the rule. Although to date most DCM genetics fits into a Mendelian rare variant disease paradigm, this paradigm may be incomplete with only 30–35% of FDC genetic cause identified. Despite this incomplete knowledge, we predict that DCM genetics will become increasingly relevant for genetics and cardiovascular professionals. This is because DCM causes heart failure, a national epidemic, with considerable morbidity and mortality. The fact that early, even pre-symptomatic intervention can prevent or ameliorate DCM, coupled with more cost-effective genetic testing, will drive further progress in the field. Ongoing questions include: whether sporadic (IDC) disease has a genetic basis, and if so, how it differs from familial disease; which gene-specific or genetic pathways are most relevant; and whether other genetic mechanisms (e.g., DNA structural variants, epigenetics, mitochondrial mutations and others) are operative in DCM. We suggest that such new knowledge will lead to novel approaches to the prevention and treatment of DCM.
Similar content being viewed by others


Main
Dilated cardiomyopathy (DCM) has recently emerged as having a genetic basis, much as did hypertrophic cardiomyopathy (HCM) in the 1990s. The discovery of genetic cause for some of DCM, otherwise thought to be idiopathic, and the rapid development of more cost-effective molecular genetic testing for rare variants bring an opportunity for collaboration between genetics professionals and cardiovascular specialists in DCM evaluation and diagnosis.
Knowledge of DCM is increasingly essential for genetics professionals in both general genetics practices staffed by clinical geneticists and genetic counselors and in cardiovascular genetic medicine clinics staffed by cardiovascular and genetic professionals.1 This is because we predict that genetic DCM will rapidly emerge from an uncommon diagnosis rarely seen in either genetics or cardiology clinics, to a mainstream genetics diagnosis, now associated with >30 genes. This prediction is based on four key facts. First, DCM of all causes underlies at least half of the heart failure epidemic in the United States, where the heart failure syndrome is defined as an inadequate cardiac output to provide circulatory and nutrient support to the body. Heart failure, from American Heart Association statistics in 2010, affected approximately 5.8 million US citizens,2 of which a significant portion will be diagnosed with DCM of unknown cause (otherwise characterized as idiopathic DCM [IDC]). Second, a genetic cause has been demonstrated for an estimated 30–35% of IDC (in familial or apparently sporadic cases), making testing feasible. Third, the recent dramatic progress with more cost-effective genetic testing makes predictive diagnosis possible and enhances presymptomatic diagnosis. Finally, and perhaps most importantly, presymptomatic interventions of DCM have proven value to prevent morbidity and mortality.
We also note that the classic Mendelian rare variant paradigm may be incomplete to characterize genetic DCM.3 Although considerable progress has been made in discovering the genetic cause of a fraction of DCM, providing an initial foothold for clinical practice, we also predict that with the availability of exome and whole genome sequencing, our understanding of DCM genetics will transition into a more complex rare variant paradigm.3 Hence, we will need genetics professionals to contribute to the DCM research effort and to help manage the clinical aspects of this important entity.
DCM: EPIDEMIOLOGY, NOMENCLATURE, AND CLINICAL CHARACTERISTICS
Definition and diagnosis of DCM
DCM is characterized by left ventricular enlargement (LVE) and systolic dysfunction with an ejection fraction (EF) < 50%,4 or, more stringently, <45% (Table 1).5 Approximately 35–40% of DCM cases are assigned a diagnosis of IDC after detectable causes have been excluded. The most common DCM cause in the United States, ischemic heart disease due to coronary artery disease (CAD), needs to be excluded in men older than 40 years and women older than 45 years (or at younger ages if risk factors are present, e.g., cigarette smoking, diabetes, hypertension, or a strong family history of early coronary disease). Less common causes of DCM that need to be excluded include structural heart disease (congenital or valvular), thyroid disease, iron overload, and exposure to cardiotoxins such as anthracyclines, chest radiation, and other much less common conditions, including those accompanying inflammatory arthritides, myocarditis (e.g., giant cell myocarditis), protozoal infections (e.g., Chagas disease), and many others (Table 2). HCM may occasionally show characteristics of DCM (reduced systolic function and some dilatation) late in its course (Table 1). Extensive literature, not reviewed in this study, is available for HCM.9–12
DCM nomenclature: IDC
DCM can be used either as a generic term to include all causes of LVE and systolic dysfunction, separating DCM from the two other classic cardiomyopathy categories, HCM or restrictive cardiomyopathy (RCM) (Table 1).11 Further, it has become common practice within heart failure clinical trials to assign patients into categories of “ischemic” or “nonischemic” DCM. The former category includes anyone with ischemic heart disease, most commonly from prior myocardial infarction and/or CAD, defined most stringently for research purposes as at least one epicardial coronary artery with >50% narrowing. However, this research standard may be too stringent for clinical (or clinical trial) purposes, as it is not uncommon to observe DCM with CAD and coronary narrowing of 50–70% (and at times involving multiple vessels) without evidence of prior myocardial infarction that may be adjudicated by cardiovascular specialists as “nonischemic cardiomyopathy with incidental CAD.” Nonischemic cardiomyopathy is used to categorize all other causes of DCM (Table 2), although the majority comprises DCM of unknown etiology. This latter category, termed IDC, is a diagnosis of exclusion. IDC is used to describe the phenotype (Table 3). Assignment of IDC requires a careful and complete medical and at least a three-generation family history, a comprehensive physical examination, an electrocardiogram, an echocardiogram, and further testing as indicated (see later).
Familial DCM
A diagnosis of familial DCM (FDC) is assigned when IDC occurs in at least two closely related family members.4,5 This is a phenotypic diagnosis (Table 3). Of these cases, we have recently shown that approximately 28% have nonsynonymous rare variants in 14 genes previously shown to cause DCM.13 Most genes implicated in genetic DCM/FDC are autosomal with dominant transmission, although a few follow an autosomal recessive, X-linked, or mitochondrial pattern of inheritance4,11,14 (Table 4).
DCM nomenclature: Genetic DCM
The genetic basis of IDC has recently emerged, and hence at this time, the terminology of “genetic DCM,” (Table 3) much less a more specific gene-based diagnosis (e.g., LMNA cardiomyopathy), is not part of the common diagnostic lexicon. However, we propose this approach (Table 3).
IDC epidemiology
IDC affects all ethnic groups. An epidemiologic study conducted in Olmsted County, Minnesota, in 1989, estimated the incidence of IDC at 6/100,000, and prevalence was estimated at 36.5 per 100,000.76 That same study found that the prevalence of HCM was 19.7 per 100,000.76 This study may have significantly underestimated both IDC and HCM prevalence, as subsequent studies have estimated HCM prevalence to be 1/500,77 (or 200/100,000, 10 times more prevalent than the Olmsted County study). From a variety of sources, it is likely that the incidence and prevalence of DCM have also been significantly underestimated. Heart failure experts suggest that IDC is at least as common as HCM, with estimates ranging up to twice that of HCM, but no further published studies are available.
LVE and systolic dysfunction
The DCM diagnosis by definition requires the presence of LVE and reduced systolic function, both most commonly evaluated by transthoracic echocardiography. The echocardiographically determined upper limits of normal of the left ventricular (LV) chamber size have historically been defined using an algorithm based on body surface area.78 More recently, echocardiographic data from 1099 normal subjects from the Framingham heart study led to more rigorous definitions of echocardiographic normals using a height- and gender-based approach,79 which has been used in our DCM research.80,81 Defining the upper limits of normal LV size is imperative for clinical interpretation, so that mildly dilated left ventricles will not be missed, particularly in females of shorter stature. Regardless of approach, genetics professionals conducting family-based evaluations of early DCM in at-risk relatives must insist on careful LV measurements in end diastole using current tables or algorithms for determining upper limits of normal. This concept, however, has not been incorporated into echocardiogram reports on a regular basis.
We note, however, that in familial studies that include cardiovascular screening of asymptomatic relatives of individuals with IDC, systolic dysfunction has been observed to precede LVE or vice versa. A key IDC/FDC phenotyping study82 observed that LVE preceded systolic dysfunction in a number of cases and proposed LVE as an early sign of DCM. In other cases, the opposite has been observed, where reduced systolic function precedes LVE.
Systolic function is almost always estimated by a measure of the LV ejection fraction, most commonly by echocardiography, nuclear studies, or cardiac magnetic resonance (CMR) imaging. Systolic function, measured by EF provided in EF percentage units, can be most accurately assessed with high reliability with nuclear studies performed in good laboratories. For example, a multiuptake gated acquisition study can provide measures of LV ejection fraction (±3% EF units) that are considered superior to that of echocardiography (±5–10% EF units). The mean EF by nuclear studies in a population of normal subjects in most laboratories is approximately 65% ± 10% representing two standard deviations. The normal EF for echocardiographic studies is also 65% ± 10%, but because of issues of precision, an echocardiographically derived EF of 50–55% is considered a gray zone, and an echocardiographic EF < 50% is considered abnormal.
Another measure of systolic function, fractional shortening (FS), is a ratio of the LV end dimension in systole (LVEDs) compared with the LV dimension in end diastole (LVEDd) that can be simply calculated from echocardiographic LV measurements (FS = [LVEDd − LVEDs]/LVEDd). A FS of <25–28% is also indicative of systolic dysfunction.
CMR, because of its superior imaging capabilities, is recently considered the gold standard for measures of chamber size, function, mass, and other parameters, and with expert interpretation, is always reasonable to consider in the initial evaluation of cardiomyopathy. However, compared with echocardiography, CMR is not universally available, CMR is more expensive and time intensive for conduct and analysis, and CMR is contraindicated for most existing pacemakers and implantable cardiac defibrillators (ICDs) because of concern of disruption of circuitry and heating of endocardial leads. However, new generations of pacemakers and ICDs will be CMR compliant. Ongoing CMR research may also provide unique parameters to help detect very early clinical disease (e.g., Ref. 83), but given the financial, accessibility, and device-related limitations of CMR, echocardiography remains the usual initial approach to clinical evaluation.
Disease presentation—IDC
Although IDC may be asymptomatic for months to years, it almost always presents late in its clinical course, usually with serious and/or life-threatening advanced disease such as heart failure, sudden cardiac death (SCD), or stroke from mural thrombus. However, early medical intervention can be highly effective to ameliorate disease and in some cases to reverse the phenotype. This provides the compelling rationale for presymptomatic diagnosis, warranting clinical and molecular screening of at-risk relatives.
Age of onset or diagnosis
IDC commonly presents in the 4th–6th decades of life; however, onset in infancy and early childhood has been reported, as well as in the elderly.11 When advanced disease presents in a proband with no prior knowledge of risk for DCM, its presentation may include heart failure with symptoms of congestion (edema, orthopnea, and paroxysmal nocturnal dyspnea) and/or reduced cardiac output (fatigue, shortness of breath, dyspnea, on exertion); arrhythmia and/or conduction system disease (CSD), including syncope or presyncope, bradycardia, tachycardia, supraventricular or ventricular arrhythmias including atrial flutter or atrial fibrillation, ventricular tachycardia (VT), or SCD; or stroke or other embolic phenomena from mural thrombus.4,84,85 DCM can also be present in asymptomatic individuals and is discovered from preventive screening or serendipitously from medical evaluation for other reasons.4,84,85
Disease presentation in FDC
The age of onset, presentation, and disease course in a proband will not necessarily be helpful to predict prognosis in other family members, even those shown to carry the same mutation.81
DCM presenting in pregnancy
DCM may also occur during pregnancy and is commonly termed peripartum cardiomyopathy (PPCM) or pregnancy-associated cardiomyopathy (PACM). We have recently shown that some women with PPCM or PACM carry rare mutations in established DCM genes.86 PPCM has been traditionally defined as DCM onset during the last month of pregnancy to 5 months postpartum. This is an arbitrary definition, and a related term, PACM, refers to onset occurring before the last month of pregnancy.87 Although considered a different clinical entity by some,87,88 we did not observe any clinical differences in the 45 cases assigned as either PACM or PPCM from our cohort of 520 families with DCM.86 Of these 45 cases, 19 had been sequenced for known DCM genes, and six of these carried a mutation in a gene previously observed to carry mutations causing DCM (MYBPC3, MYH7, TNNT2, SCN5A, MYH6, and PSEN2). All 45 cases were embedded in FDC/IDC families.86
DCM with CSD that may be associated with prominent arrhythmias
A subset of individuals with DCM also exhibit CSD and/or cardiac arrhythmias,81 particularly those with LMNA or SCN5A mutations (Table 4). In a subset of cases, CSD with or without arrhythmia may be the presenting clinical feature, and in some families with a known genetic cause, individuals have been shown to exhibit only CSD and arrhythmia.56 Family studies (see references in Table 4) suggest that CSD commonly precedes the development of DCM in these individuals by a few years to more than a decade.
CSD includes first, second, or third degree heart block, usually identified by a resting electrocardiogram (ECG) or bundle branch blocks.56 CSD involvement commonly starts with disease of the sinus node and/or atrioventricular node that can manifest as sinus bradycardia, sinus node arrest with junctional rhythms, or heart block.89 Associated cardiac arrhythmias include the presence of paroxysmal or sustained atrial flutter or fibrillation, paroxysmal or sustained supraventricular arrhythmias, symptomatic brady-tachy syndrome (sick sinus syndrome), VT or ventricular fibrillation (VF), or resuscitated SCD.56
The use of a permanent pacemaker or ICD is indicated for CSD or to prevent or treat lethal cardiac arrhythmias. When taking a family history, the presence of a pacemaker or ICD in relatives can be helpful to indicate cardiovascular disease.56 SCD due to arrhythmia tends to occur with progressive disease, although SCD as the presenting manifestation has been reported in LMNA-related DCM,89 as well as other DCM genetic etiologies, particularly from SCN5A-related DCM (see references in Table 4).
Nuances of the term “sudden cardiac death” and implications for family history
We clarify in this review that the SCD term in the cardiovascular literature does not necessarily imply a vital status outcome of the subject, that is, an individual who has suffered SCD may remain alive, having been resuscitated and his/her rhythm successfully treated. In this way, the SCD term denotes either a lethal life-threatening arrhythmia (usually fast VT or VF) that required medical intervention for survival or the occurrence of death of the individual, suddenly, that has been attributed to cardiovascular cause (and in the clinical trial literature, in most cases, arrhythmic death). We also clarify that when taking a cardiovascular family history, relatives can “die suddenly” from acute myocardial infarction, which in almost all cases results from VT or VF associated with the myocardial ischemia occurring during the acute myocardial infarction, triggering the arrhythmia. Although this death technically results from arrhythmia as a consequence of an acute event from CAD, and although such history may be informative for other genetic and/or familial causes of coronary atherosclerosis, including familial hypercholesterolemia, it is not useful for purposes of ascertaining whether the subject has genetic DCM because of its fundamentally different etiology. On the other hand, the incidence and prevalence of CAD are high in a first world population, and therefore, it is not uncommon for a DCM gene carrier to also have CAD, confounding family history assessments. Because of the age dependency of CAD, we treat any unexpected death of men younger than 40 years and women younger than 45 years as suspicious for DCM. For those older individuals who had an episode of SCD or died suddenly of cardiac cause, CAD needs to be ruled out for the SCD to be suggestive of an underlying DCM mutation.
Syndromic DCM
Although most genetic DCM involves only the heart, a number of syndromic genetic conditions include DCM as a feature. Selected examples are provided (Table 5). For this reason, careful attention should be paid to the family history and physical examination to rule out syndromic disease. In most cases of syndromic disease involving DCM, multiple tissues and/or organ systems are involved, the most common of which is skeletal muscle.11 The ratio of syndromic compared with nonsyndromic DCM is unknown.
DCM GENETICS
Clinical genetics
Cardiovascular screening in family members of IDC probands has revealed FDC in 20–35% of cases.82,96–99 Using LVE alone as an early indicator of IDC, familial disease has been found in up to 48% of probands.82 In general, the clinical cardiovascular characteristics of those with IDC6 and those with FDC do not differ.4,5,81,84,100
Estimates suggest that IDC is inherited in an autosomal dominant pattern in about 90% of kindreds and shows reduced, age-dependent penetrance and variable expressivity.4,5,11,81,84,96,100 Exact penetrance estimates are not available, although one study calculated 90% penetrance after the age of 40 years in autosomal dominant forms99 and as high as 100% in LMNA mutation carriers older than 30 years.101 Autosomal recessive and X-linked inheritance has also been reported in 1–2% and 5–10% of cases, respectively.14
Molecular genetics of IDC/FDC: Marked locus heterogeneity
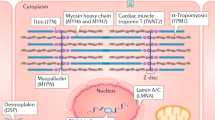
FDC is a genetically heterogeneous disease. More than 30 genes, almost all autosomal, encoding proteins of diverse roles have been identified (Table 4), demonstrating a degree of locus heterogeneity that is comparable with few other genetic disorders. Genetic DCM differs from HCM and arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C) most notably in the number of genes associated with disease and the diversity of gene/protein function. For instance, in HCM, two genes, MYH7 and MYBPC3, account for 40–60% of cases or 80–90% of genetic cause when a genetic basis can be established.102,103 In ARVD/C, three genes, PKP2 (plakophilin 2), DSP (desmoplakin), and DSG2 (desmoglein 2) account for 40–50% of cases.104,105 Further, although mutations in genes encoding proteins of diverse function have been identified in FDC, most HCM and ARVD/C mutations occur in genes encoding sarcomeric or desmosomal proteins, respectively (see Refs. 11 and 14). A predominant LV ARVD/C phenotype has also been described that presents with prominent arrhythmias that can present phenotypically as DCM.106,107
To date, a subset of 14 genes, screened by bidirectional Sanger sequencing for mutations, collectively accounted for approximately 28% of likely or possible cause in a series of 312 probands with IDC/FDC.13 The fraction of cause of DCM attributed to nonsynonymous, splice site, and small insertion/deletion mutations in coding regions and intron/exon boundaries of these genes is shown (Table 4). The remainder of the genetic cause has been estimated from single gene reports in various cohorts (Table 4). These data indicate that, to date, mutations in LMNA, MYH7, MYBPC3, TNNT2, SCN5A, and MYH6 account for approximately 75% of known genetic cause of DCM.13 From all publications, we estimate that approximately 30–35% of DCM genetic cause has been identified (Table 4).
DCM shows marked allelic heterogeneity
Numerous mutations have been identified in FDC. Unlike cystic fibrosis, in which, despite large allelic heterogeneity, 70% of cases are caused by the [Delta]F508 mutation,85 almost all DCM mutations are private. Although there are some mutations that have been reported in multiple families, such as a TNNT2 lysine210 deletion (see references in Table 4), the vast majority of DCM mutations are private to a given family.13,25,29,30,56,57 Although most variants are missense, a few nonsense, small insertion/deletions, and splice site variants have also been reported.25,56,108 Exceptions to date include a 2 kb deletion in the EYA4 gene46 and a large LMNA deletion.109
Mitochondrial disease
Mitochondrial mutations, because of the high energy requirements of myocardium and the key role of mitochondria to synthesize ATP, have been hypothesized to cause DCM. Although ascertaining whether mtDNA mutations actually cause DCM has been challenging, some mtDNA mutations have been reported in DCM.90–95 Syndromic disease associated with DCM resulting from mitochondrial mutations (e.g., Kearns-Sayre syndrome, OMIM, #530000) has also been recognized (Table 5). We are not yet aware of next generation sequencing technologies that have been used to rigorously evaluate heteroplasmic mitochondrial DNA variation in large cohorts of patients with DCM or the evaluation of genomic sequences encoding mitochondrial proteins for nonsynonymous variants, although commercial testing panels that include mitochondrial DNA are currently available.
IDC/FDC pathology and pathogenesis: Multilocus pathways resulting in a final common phenotype
Despite dramatic locus and allelic heterogeneity, a relatively homogeneous DCM phenotype results. Histologic findings of IDC include the relatively nonspecific findings of myocyte hypertrophy, myocyte loss, and interstitial fibrosis.110 DCM has been proposed to result from a “final common pathway.”111 Alternatively, we have proposed a multilocus pathway hypothesis, where DCM is a final phenotype resulting from a variety of different disease genes or genetic injury pathways.13 In this paradigm, DCM may also result from a variety of other nongenetic causes noted earlier, such as ischemic cardiomyopathy. Because of the marked locus heterogeneity involving multiple gene networks, this hypothesis suggests that many different genetic pathways may lead to DCM. This multilocus pathway hypothesis is relevant because if many different disease genes or gene networks cause DCM, then specific interventions, targeted to these genes or gene cascades, may be relevant for therapy. Testing this hypothesis will require identifying and characterizing these genetically active pathways or gene cascades in DCM.
Genotype/phenotype correlations
No clear genotype-phenotype correlations exist in genetic DCM. Possible exceptions, as noted earlier, include the prominent CSD and arrhythmia that precede the onset of LVE and/or systolic dysfunction for LMNA and SCN5A mutations. Particularly for LMNA, the CSD consists of heart block, sustained atrial flutter/fibrillation, paroxysmal supraventricular arrhythmias, atrial flutter/fibrillation, symptomatic brady-tachy syndrome, VT/fibrillation, and SCD.4,11,56 LMNA cardiomyopathy may also show relatively less LVE compared with the degree of systolic dysfunction, particularly early in its course (see references in Table 4). SCN5A mutations also commonly present with CSD during adolescence progressing to LV dysfunction.69
We25,29,108 and others30,26,27,31 have also observed that TNNT2 mutations are commonly associated with DCM of early and aggressive onset, often in the 2nd and 3rd decades of life. Despite this, we have also more recently documented disease in a 70-year-old woman with onset at the age of 69 years, who was found to have a TNNT2 mutation shown to be likely pathogenic through functional studies (Morales et al., unpublished data). This illustrates the uncertainties of relying on any specific phenotype/genotype association in genetic DCM.
Phenotypic heterogeneity and plasticity
Phenotypic heterogeneity has been observed for several genes associated with DCM. LMNA mutations have shown dramatic phenotypic heterogeneity (e.g., Hutchison-Gilford progeria, partial lipodystrophy, mandibular acral dysplasia, Charcot-Marie-Tooth type 2B1, and others),112 with skeletal myopathy and CSD with or without DCM the most relevant to this topic. Other heterogeneity has been observed in genes encoding sarcomeric proteins, most commonly with DCM or HCM from mutations in MYH7,25 TNNT2,25,32,113 or MYBPC3.13 One multiplex family showed DCM, HCM, and RCM phenotypes in different family members harboring a single mutation in TNNT2.113
“Apparently sporadic” IDC
Most genetic discovery research in the 1990s and early 2000s targeted large FDC kindreds. Thus, the extent of genetic causation in sporadic IDC remains an open question. We emphasize the term “apparently sporadic IDC” to mean phenotype studies where the available data were derived from probands reporting a negative family history, as their relatives were not systematically screened clinically (echocardiogram and ECG) to rule out DCM. Ruling out FDC by clinical cardiac screening, especially imaging studies to assess LV size and function, is essential given that early signs of DCM, even to the extent of meeting full criteria for IDC, can be present in completely asymptomatic individuals. We again emphasize that the family history is known to be insensitive to detect FDC and that a family member may have a completely normal history, examination, and ECG but still be affected when an echocardiogram or equivalent cardiac imaging is performed.
The currently available studies of systematic resequencing in sporadic IDC cohorts are limited. To date, no major prospective study (including our prior work in this area13,25,48,54,56) has systematically resequenced the most common DCM genes in a cohort of sporadic IDC patients where first-degree relatives were screened clinically (history, examination, ECG, and echocardiogram) to rule out familial DCM. Despite this caveat, our resequencing studies suggest that apparently sporadic IDC may also have a genetic basis.13,25,54,56 For example, in our LMNA resequencing study, we identified mutations in 3.6% of apparently sporadic IDC and 7.5% of FDC.56 Further, our resequencing study of six genes (MYH7, TNNT2, SCN5A, CSRP3, LBD3, and TCAP) in 313 DCM probands identified mutations in 10.8% with apparently sporadic IDC and in 9.8% with FDC.25 A follow-up resequencing study of five additional genes (MYBPC3, MYH6, TPM1, TNNC1, and TNNI3) in 312 subjects (311 from the previous cohort and one additional case) identified mutations in 9.2% with apparently sporadic IDC and in 11.6% with FDC.13 Confirming that sporadic DCM has a genetic basis will require formal studies with clinical screening to confirm sporadic DCM, followed by comprehensive genetic studies.
MANAGEMENT APPROACHES TO DCM
Treatment
Treatment of individuals with symptomatic DCM is recommended, per American Heart Association/American College of Cardiology guidelines114,115 for DCM and heart failure: in brief, ACE-inhibitors, beta blockers, and ICDs when indicated.114,115 Guidelines for genetic cardiomyopathies have also been recently prepared by the Heart Failure Society of America.11 For patients with advanced heart failure, the above measures in addition to diuretics and inotropes, as indicated, and for end-stage disease, consideration of ventricular assist devices and/or cardiac transplantation.114,116 Treatment of asymptomatic LV dysfunction from any cause with beta blockers and ACE-inhibitors will delay onset of symptoms, improve LV function, and will likely improve mortality.14,114 Formal studies have not yet been completed with asymptomatic and very early genetic DCM, but most cardiovascular experts suggest that treatment will likely improve outcomes.117 Resolution of this question is one that deserves carefully designed gene-specific clinical trials.
Management of asymptomatic CSD in someone with suspected or proven FDC requires close surveillance. At times, progressive but asymptomatic CSD in family members harboring LMNA mutations may require prophylactic pacemaker or ICD placement, an issue that requires expert electrophysiological consultation in concert with other cardiovascular and genetics assessments. This important issue has been noted in the 2009 Heart Failure Society of America guideline document.14
Genetic counseling
As per guidelines, genetic counseling is recommended for all patients and families with cardiomyopathy,14 particularly in light of the complexity and rapidly evolving options for genetic testing in DCM. Multidisciplinary medical care involving genetic counselors and cardiologists, medical geneticists, or other experts in the field of cardiovascular genetics is ideal.
Genetic counseling includes obtaining a careful and comprehensive family history, education regarding disease transmission, advice on potential risks and benefits of cardiac screening and genetic testing, assistance in interpreting genetic test results, and helping patients and their families adapt to the psychosocial consequences of genetic disease.14,84,100
A targeted three- to four-generation pedigree is essential in the medical evaluation of DCM.4,14,84,100 Key questions include age at the onset of symptoms, as IDC onset at a young age is suggestive of a strong genetic component.84,100 Infantile onset of cardiomyopathy is often consistent with autosomal recessive, X-linked, or mitochondrial inheritance.4,14,84,100 Inquiry is important regarding symptoms of heart failure (edema, orthopnea, paroxysmal nocturnal dyspnea, dyspnea on exertion, and fatigue) and arrhythmia (palpitations, presyncope, syncope, and SCD).4,14,84,100 Symptoms of heart failure in the peripartum period can be indicative of PPCM86,118 and should also be noted.
Documentation of SCD, unexplained death at a young age, and history of other heart conditions such as “heart attack” (myocardial infarction) should also be documented. This attribution (“heart attack”) is commonly used by many patients to describe any CV condition that caused a CV hospitalization in a family member. Follow-up questions to ascertain whether the family member in question underwent stent placement or had a coronary artery bypass grafting operation can help identify ischemic versus nonischemic disease. Results from other tests and procedures such as a coronary angiogram, echocardiogram, multiuptake-gated acquisition, electrocardiogram, 24-hour ECG recording (commonly referred to as a “Holter monitor”), endocardial biopsy, and others should be documented.
Assessment for possible syndromic forms of DCM (Table 5) requires careful attention to the history and examination. For example, noting sensorineural hearing loss, which may be observed in people with FDC and an EYA4 mutation should be noted (Table 4). DCM can be a part of the spectrum of disease for several mitochondrial syndromes.11 Muscle weakness and elevated CPK levels suggests DMD-related Duchenne or Becker muscular dystrophy or LMNA-associated myopathy. Short stature, neutropenia, and congenital DCM suggest Barth syndrome. Findings with LMNA-DCM associated with Emery-Dreifuss muscular dystrophy include skeletal muscle weakness, contractures, a waddling gait, and toe walking among others.100 Basic knowledge of signs and symptoms of other types of cardiomyopathy, particularly HCM (Table 1), and other arrhythmia syndromes such as the Long QT syndrome, is also important, as symptoms and at times genetic cause can overlap with those seen in FDC.100
Clinical cardiovascular screening of relatives
Clinical screening is recommended for first-degree relatives of probands with IDC.14 The standard for cardiac screening in those at risk for DCM includes a careful medical and family history, an echocardiogram, an electrocardiogram, and a physical examination. If any cardiovascular abnormalities are detected, a full cardiovascular evaluation by a cardiovascular specialist is indicated. This is particularly relevant when symptoms or signs of DCM are identified in those whose coronary disease risk is increasing (men older than 40 years, women older than 45 years, modified to earlier ages with added coronary risk factors such as cigarette smoking, diabetes, hypertension, a positive family history of early CAD, or hyperlipidemia). CMR mentioned earlier can also be useful, especially for questions of overlap of DCM with HCM, RCM, or ARVD/C.
Screening asymptomatic relatives
It is imperative to understand that genetic DCM can occur in individuals who are completely asymptomatic. Presymptomatic diagnosis of FDC emphasizes the importance of preventive screening in first-degree relatives of individuals with IDC.14 Presymptomatic diagnosis allows for early intervention and may result in the prevention or delayed onset of heart failure or SCD.14
Screening intervals for asymptomatic relatives
Regardless of genetic testing status in the family (i.e., an established mutation, genetic testing that is negative or has shown a variant of unknown significance, or no genetic testing), asymptomatic relatives with any evidence suggestive of DCM on cardiac screening studies should be rescreened on a regular basis.14 This group should be followed up by cardiovascular specialists with expertise in DCM diagnosis and treatment.
Follow-up for asymptomatic individuals who carry a putative disease-causing DCM mutation but who have thus far had negative clinical cardiac screening is recommended yearly in childhood and then every 1–3 years in adulthood.14 For families in which a mutation has not been identified in a proband, relatives with negative clinical cardiac screening should be rescreened every 3–5 years beginning in childhood.14 Similarly, for families with a family history of IDC in which no genetic testing has been pursued, individuals with negative cardiac screening should seek rescreening every 3–5 years.14
At this time, continued screening has not been recommended14 for those family members who test negative for their family's DCM-causing mutation. However, they should be informed of the symptoms associated with DCM (arrhythmia, heart failure, etc.) and should be instructed to bring such symptoms promptly to medical attention should they occur. In relatives who do not carry the putative family DCM mutation, the residual risk of developing DCM has not yet been assessed, but the marked locus heterogeneity and the estimate that only 30–35% of genetic cause has been identified for DCM make this a concern.
Genetic testing guidelines
Clinical genetic testing for DCM has been evolving rapidly, from testing one or two genes, such as LMNA, at selected laboratories, on a gene-by-gene basis, to panels containing 10–30 genes. The recently published guidelines (2009) for the evaluation of genetic cardiomyopathies14 recommended a stepwise genetic testing process, beginning with the three genes accounting for approximately half of DCM risk (LMNA, TNNT2, and MYH7) and, if negative, reflex to the remainder of genes available for testing.14 These guidelines have already become outdated, as molecular genetic testing laboratories now offer DCM genetic testing panels of 12–30 genes using next generation sequencing methods.
Testing approach within a family
Following family-based genetic testing principles, genetic testing should begin with an affected family member available for testing and ideally the one who is the most severely affected. In addition to the common genes associated with DCM, the large, currently available genetic panels also include syndromic disease genes and genes with limited association data with DCM. This reinforces the emphasis on a careful physical examination and family history to rule out syndromic disease. Pretest genetic counseling should also include a thorough discussion of the likelihood of finding a variant of unknown significance (VUS), the meaning of a VUS, and the need for cooperation among family members to assess the pathogenicity of a VUS. Because a genetic cause is likely to be identified in only 15–25% of cases, genetic testing of an unaffected family member has not been recommended. If an affected family member is unavailable, investigations should be conducted to identify stored tissue suitable for DNA analysis from an affected, deceased family member. In the absence of an affected relative or stored tissue, testing of an unaffected family member may be considered with expert counseling and communication of the limitations of this testing approach. Insurance coverage is also variable and can be a limiting factor in deciding where to begin testing in the family.11,14
FUTURE DIRECTIONS: A MORE COMPLETE MODEL FOR DCM GENETICS?
Limitations of current DCM genetic studies
Current research has only scratched the surface of DCM genetics. Most recent efforts have been devoted to DCM gene discovery or to preliminary studies of mutation frequency and character in modest sized DCM cohorts, with almost exclusive focus on mutation detection in coding sequence. We estimate that only 30–35% of DCM genetic cause has been identified. The limited research into mutations of genomic or mitochondrial DNA encoding mitochondrial proteins has been noted earlier. No systematic study of structural variants has been published for any DCM cohort. Although our resequencing studies13,25 have examined some intronic and 5′- and 3′-untranslated regions in 11 genes (unpublished data), in general such noncoding sequence has not been reported in DCM gene studies. Similarly, the search for rare variants in near or far upstream regions harboring promoter sequences have not been reported. We also note the lack of investigation of epigenetic mechanisms that may also be key to understanding DCM genetics.
Molecular research has been framed within a Mendelian, rare variant paradigm disregarding common variants, although a recent study119 of our first resequencing dataset25 suggested that such analyses were feasible and may contribute to understanding potential genetic modifiers. Common polymorphisms in DCM genes may be highly relevant for disease expression, as illustrated by SCN5A variants affecting SCN5A disease-causing mutations (Cheng et al., unpublished data). These issues will need to be addressed with exome or whole genome sequence analysis in DNA specimens from hundreds of IDC and FDC probands.
A complex rare variant DCM genetic disease model
DCM genetics has been viewed through the lens of Mendelian genetics, specifically where high penetrance mutations in a few genes cause disease.3 Most of the published evidence (Table 4) supports this model and particularly for highly penetrant nonsynonymous variants in sarcomeric proteins (e.g., TNNT2 and MYH7). However, aspects of our resequencing data, although quite preliminary, suggest a more complex genetic DCM model.3 In our LMNA resequencing study, at least one individual with DCM in 6 of 19 (32%) FDC kindreds with a putative disease-causing LMNA mutation showed nonsegregation, that is, at least one affected family member was negative for the family mutation.56 These findings suggest that in DCM, multiple mutations may be at play more commonly than anticipated, perhaps underlying penetrance, expressivity, or even causation. Additional complexity is possible in that multiple rare (allele frequencies <0.001–0.005) or “not so rare” variants (allele frequencies of 0.002–0.01) may be relevant.3 These issues have also been recently explored for other conditions.120,121
A related question is whether familial or sporadic IDC lie on a genetic continuum, resulting from different degrees of genetic influence. A corollary of this question is the fact that the genetic factors underlying penetrance and variable expressivity in FDC are still unknown. The genetic basis of sporadic IDC remains to be determined, whether from rare, single, de novo high penetrance mutations or from the cumulative effects of more than one rare, moderate to low penetrance variants. In our preliminary studies, we have observed that approximately 3% of our DCM probands (our unpublished data), when resequenced for multiple genes,13,25 have multiple rare nonsynonymous variants. Similar findings have been observed in other cardiovascular genetic phenotypes, including 5% of HCM,102,103,122,123 5–8% of the long QT syndrome,124,125 and in ARVD/C.126 However, unlike HCM, the long QT syndrome, and ARVD/C, where approximately 65%, 75%, and 50% of genetic cause has already been identified, respectively, we estimate that only approximately 25–30% of genetic cause has been found in DCM spanning more than 30 genes. Hence, we suggest that it may be possible that rare variants in many additional genes may be relevant to cause or modify the DCM phenotype. It has been postulated that most rare missense mutations may be deleterious in humans,127 and thus far, the available data show that IDC arises from rare variants, which remain the foundation for interpreting DCM genetic data.121,128 Therefore, an oligogenic model120 may more appropriately describe some aspects of DCM than a Mendelian model.3,120,121 Other genetic models are possible, especially those informed from untapped research areas (e.g., structural variants, 3′- or 5′-untranslated regions, and epigenetics).
Exome sequencing129,130 now and whole genome sequencing soon will provide the basis to identify additional variants, both rare and common, that may act in concert to modulate the DCM phenotype. Functional studies (e.g., strengthening association with disease, for examples, see Refs. 108 and 131 and Morales et al., submitted) of such variants will be imperative to draw firm conclusions.
CONCLUSION
Although a great deal of progress for IDC and FDC has been made in discovering genetic cause and providing guidelines for its management, much more extensive research is needed, including genetic discovery and medical management of specific types of genetic DCM. As important, awareness that IDC and FDC are conditions with significant underlying genetic etiology is imperative for both the genetics and cardiovascular communities. Much greater understanding of DCM genetics will be required, including discovery of the remaining major portion of genetic cause and determining the frequency and spectrum of multiple mutations. Resolution on the issue of whether sporadic (IDC) disease has a genetic basis, and if so, how it differs from familial disease is also key. Finally, we hope that new strategies, including those devoted to gene-specific or genetic pathways, will lead to novel approaches to the prevention and treatment of DCM.
References
Hershberger RE . Cardiovascular genetic medicine: evolving concepts, rationale and implementation. J Cardiovasc Trans Res 2008; 1: 137–143.
Lloyd-Jones D, Adams RJ, Brown TM, et al. Heart disease and stroke statistics—2010 update. A report from the American Heart Association. Circulation 2010; 121: e46–e215.
Hershberger RE . A glimpse into multigene rare variant genetics: triple mutations in hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 55: 1454–1455.
Burkett EL, Hershberger RE . Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol 2005; 45: 969–981.
Mestroni L, Maisch B, McKenna W, et al. Guidelines for the study of familial dilated cardiomyopathies. Eur Heart J 1999; 20: 93–102.
Dec G, Fuster V . Idiopathic dilated cardiomyopathy. N Engl J Med 1994; 331: 1564–1575.
Manolio TA, Baughman KL, Rodeheffer R, et al. Prevalence and etiology of idiopathic dilated cardiomyopathy (summary of a National Heart, Lung, and Blood Institute workshop [see comments]). Am J Cardiol 1992; 69: 1458–1466.
Taylor MR, Carniel E, Mestroni L . Cardiomyopathy, familial dilated. Orphanet J Rare Dis 2006; 1: 27.
Maron BJ . Hypertrophic cardiomyopathy: a systematic review. JAMA 2002; 287: 1308–1320.
Maron BJ, McKenna WJ, Danielson GK, et al. American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J Am Coll Cardiol 2003; 42: 1687–1713.
Hershberger RE, Cowan J, Morales A, Siegfried JD . Progress with genetic cardiomyopathies: screening, counseling, and testing in dilated, hypertrophic, and arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Heart Fail 2009; 2: 253–261.
Cirino AL, Ho C . Familial hypertrophic cardiomyopathy overview. In: GeneReviews at GeneTests: Medical Genetics Information Resource (database online), 2008. Available at: http://www.genetests.org. Accessed August 16, 2010.
Hershberger R, Norton N, Morales A, et al. Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1 And TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circ Cardiovasc Genet 2010; 3: 155–161.
Hershberger RE, Lindenfeld J, Mestroni L, et al. Genetic evaluation of cardiomyopathy—a Heart Failure Society of America practice guideline. J Card Fail 2009; 15: 83–97.
Olson TM, Michels VV, Thibodeau SN, Tai YS, Keating MT . Actin mutations in dilated cardiomyopathy, a heritable form of heart failure. Science 1998; 280: 750–752.
Mayosa B, Khogali S, Zhang B, Watkins H . Cardiac and skeletal actin gene mutations are not a common cause of dilated cardiomyopathy. J Med Genet 1999; 36: 796–797.
Takai E, Akita H, Shiga N, et al. Mutational analysis of the cardiac actin gene in familial and sporadic dilated cardiomyopathy. Am J Med Genet 1999; 86: 325–327.
Tesson F, Sylvius N, Pilotto A, et al. Epidemiology of desmin and cardiac actin gene mutations in a european population of dilated cardiomyopathy [In Process Citation]. Eur Heart J 2000; 21: 1872–1876.
Zolty R, Brodsky G, Perryman B, Bristow M, Mestroni L . Epidemiology of cardiac actin gene mutations in dilated cardiomyopathy. J Card Fail 1999; 5( suppl 1): 23.
Taylor MR, Slavov D, Ku L, et al. Prevalence of desmin mutations in dilated cardiomyopathy. Circulation 2007; 115: 1244–1251.
Li D, Tapscoft T, Gonzalez O, et al. Desmin mutation responsible for idiopathic dilated cardiomyopathy. Circulation 1999; 100: 461–464.
Karkkainen S, Miettinen R, Tuomainen P, et al. A novel mutation, Arg71Thr, in the delta-sarcoglycan gene is associated with dilated cardiomyopathy. J Mol Med 2003; 81: 795–800.
Tsubata S, Bowles KR, Vatta M, et al. Mutations in the human delta-sarcoglycan gene in familial and sporadic dilated cardiomyopathy. J Clin Invest 2000; 106: 655–662.
Sylvius N, Duboscq-Bidot L, Bouchier C, et al. Mutational analysis of the beta- and delta-sarcoglycan genes in a large number of patients with familial and sporadic dilated cardiomyopathy. Am J Med Genet 2003; 120A: 8–12.
Hershberger RE, Parks SB, Kushner JD, et al. Coding sequence mutations identified in MYH7, TNNT2, SCN5A, CSRP3, LBD3, and TCAP from 313 patients with familial or idiopathic dilated cardiomyopathy. Clin Transl Sci 2008; 1: 21–26.
Kamisago M, Sharma SD, DePalma SR, et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med 2000; 343: 1688–1696.
Villard E, Duboscq-Bidot L, Charron P, et al. Mutation screening in dilated cardiomyopathy: prominent role of the beta myosin heavy chain gene. Eur Heart J 2005; 26: 794–803.
Daehmlow S, Erdmann J, Knueppel T, et al. Novel mutations in sarcomeric protein genes in dilated cardiomyopathy. Biochem Biophys Res Commun 2002; 298: 116–120.
Hanson E, Jakobs P, Keegan H, et al. Cardiac troponin T lysine-210 deletion in a family with dilated cardiomyopathy. J Card Fail 2002; 8: 28–32.
Mogensen J, Murphy RT, Shaw T, et al. Severe disease expression of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy. J Am Coll Cardiol 2004; 44: 2033–2040.
Li D, Czernuszewicz GZ, Gonzalez O, et al. Novel cardiac troponin T mutation as a cause of familial dilated cardiomyopathy. Circulation 2001; 104: 2188–2193.
Moller DV, Andersen PS, Hedley P, et al. The role of sarcomere gene mutations in patients with idiopathic dilated cardiomyopathy. Eur J Hum Genet 2009; 17: 1241–1249.
Olson TM, Kishimoto NY, Whitby FG, Michels VV . Mutations that alter the surface charge of alpha-tropomyosin are associated with dilated cardiomyopathy. J Mol Cell Cardiol 2001; 33: 723–732.
Lakdawala NK, Dellefave L, Redwood CS, et al. Familial dilated cardiomyopathy caused by an alpha-tropomyosin mutation: the distinctive natural history of sarcomeric dilated cardiomyopathy. J Am Coll Cardiol 2010; 55: 320–329.
Gerull B, Gramlich M, Atherton J, et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat Genet 2002; 14: 14.
Olson TM, Illenberger S, Kishimoto NY, et al. Metavinculin mutations alter actin interaction in dilated cardiomyopathy. Circulation 2002; 105: 431–437.
Knoll R, Hoshijima M, Hoffman HM, et al. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell 2002; 111: 943–955.
Mohapatra B, Jimenez S, Lin JH, et al. Mutations in the muscle LIM protein and alpha-actinin-2 genes in dilated cardiomyopathy and endocardial fibroelastosis. Mol Genet Metab 2003; 80: 207–215.
Schmitt JP, Kamisago M, Asahi M, et al. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science 2003; 299: 1410–1413.
Haghighi K, Kolokathis F, Pater L, et al. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest 2003; 111: 869–876.
Vatta M, Mohapatra B, Jimenez S, et al. Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction. J Am Coll Cardiol 2003; 42: 2014–2027.
Carniel E, Taylor MR, Sinagra G, et al. Alpha-myosin heavy chain: a sarcomeric gene associated with dilated and hypertrophic phenotypes of cardiomyopathy. Circulation 2005; 112: 54–59.
Bienengraeber M, Olson TM, Selivanov VA, et al. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat Genet 2004; 36: 382–387.
Hayashi T, Arimura T, Itoh-Satoh M, et al. Tcap gene mutations in hypertrophic cardiomyopathy and dilated cardiomyopathy. J Am Coll Cardiol 2004; 44: 2192–2201.
Murphy RT, Mogensen J, Shaw A, et al. Novel mutation in cardiac troponin I in recessive idiopathic dilated cardiomyopathy. Lancet 2004; 363: 371–372.
Schonberger J, Wang L, Shin JT, et al. Mutation in the transcriptional coactivator EYA4 causes dilated cardiomyopathy and sensorineural hearing loss. Nat Genet 2005; 37: 418–422.
Taylor MR, Slavov D, Gajewski A, et al. Thymopoietin (lamina-associated polypeptide 2) gene mutation associated with dilated cardiomyopathy. Hum Mutat 2005; 26: 566–574.
Li D, Parks SB, Kushner JD, et al. Mutations of presenilin genes in dilated cardiomyopathy and heart failure. Am J Hum Genet 2006; 79: 1030–1039.
Inagaki N, Hayashi T, Arimura T, et al. Alpha B-crystallin mutation in dilated cardiomyopathy. Biochem Biophys Res Commun 2006; 342: 379–386.
Arola AM, Sanchez X, Murphy RT, et al. Mutations in PDLIM3 and MYOZ1 encoding myocyte Z line proteins are infrequently found in idiopathic dilated cardiomyopathy. Mol Genet Metab 2007; 90: 435–440.
Duboscq-Bidot L, Xu P, Charron P, et al. Mutations in the Z-band protein myopalladin gene and idiopathic dilated cardiomyopathy. Cardiovasc Res 2008; 77: 118–125.
Knoll R, Postel R, Wang J, et al. Laminin-alpha4 and integrin-linked kinase mutations cause human cardiomyopathy via simultaneous defects in cardiomyocytes and endothelial cells. Circulation 2007; 116: 515–525.
Duboscq-Bidot L, Charron P, Ruppert V, et al. Mutations in the ANKRD1 gene encoding CARP are responsible for human dilated cardiomyopathy. Eur Heart J 2009; 30: 2128–2136.
Li D, Morales A, Gonzalez Quintana J, et al. Identification of novel mutations In RBM20 in patients with dilated cardiomyopathy. Clin Trans Sci 2010; 3: 90–97.
Brauch KM, Karst ML, Herron KJ, et al. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J Am Coll Cardiol 2009; 54: 930–941.
Parks SB, Kushner JD, Nauman D, et al. Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Am Heart J 2008; 156: 161–169.
Fatkin D, MacRae C, Sasaki T, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med 1999; 341: 1715–1724.
Brodsky G, Muntoni F, Miocic S, et al. Lamin A/C gene mutation associated with dilated cardiomyopathy with variable skeletal muscle involvement. Circulation 2000; 101: 473–476.
Becane HM, Bonne G, Varnous S, et al. High incidence of sudden death with conduction system and myocardial disease due to lamins A and C gene mutation. Pacing Clin Electrophysiol 2000; 23: 1661–1666.
Jakobs PM, Hanson E, Crispell KA, et al. Novel lamin A/C mutations in two families with dilated cardiomyopathy and conduction system disease. J Card Fail 2001; 7: 249–256.
Arbustini E, Pilotto A, Repetto A, et al. Autosomal dominant dilated cardiomyopathy with atrioventricular block: a lamin A/C defect-related disease. J Am Coll Cardiol 2002; 39: 981–990.
Hershberger RE, Hanson E, Jakobs PM, et al. A novel lamin A/C mutation in a family with dilated cardiomyopathy, prominent conduction system disease, and need for permanent pacemaker implantation. Am Heart J 2002; 144: 1081–1086.
Taylor MR, Fain PR, Sinagra G, et al. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J Am Coll Cardiol 2003; 41: 771–780.
Sebillon P, Bouchier C, Bidot LD, et al. Expanding the phenotype of LMNA mutations in dilated cardiomyopathy and functional consequences of these mutations. J Med Genet 2003; 40: 560–567.
MacLeod HM, Culley MR, Huber JM, McNally EM . Lamin A/C truncation in dilated cardiomyopathy with conduction disease. BMC Med Genet 2003; 4: 4.
Sylvius N, Bilinska ZT, Veinot JP, et al. In vivo and in vitro examination of the functional significances of novel lamin gene mutations in heart failure patients. J Med Genet 2005; 42: 639–647.
Pethig K, Genschel J, Peters T, et al. LMNA mutations in cardiac transplant recipients. Cardiology 2005; 103: 57–62.
Karkkainen S, Reissell E, Helio T, et al. Novel mutations in the lamin A/C gene in heart transplant recipients with end stage dilated cardiomyopathy. Heart 2006; 92: 524–526.
McNair WP, Ku L, Taylor MR, et al. SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation 2004; 110: 2163–2167.
Olson TM, Michels VV, Ballew JD, et al. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA 2005; 293: 447–454.
Hershberger RE, Kushner JK, Parks SP Dilated cardiomyopathy overview In: GeneReviews at GeneTests: Medical Genetics Information Resource (database online), 2007. July 10, 2008; initial posting, July 27, 2007. Available at: http://www.genetests.org. Accessed August 16, 2010.
Towbin JA, Hejtmancik JF, Brink P, et al. X-linked dilated cardiomyopathy. Molecular genetic evidence of linkage to the Duchenne muscular dystrophy (dystrophin) gene at the Xp21 locus. Circulation 1993; 87: 1854–1865.
Muntoni F, Cau M, Ganau A, et al. Brief report: deletion of the dystrophin muscle-promoter region associated with x-linked dilated cardiomyopathy. N Engl J Med 1993; 329: 921–925.
D'Adamo P, Fassone L, Gedeon A, et al. The x-linked gene G4.5 is responsible for different infantile dilated cardiomyopathies. Am J Hum Genet 1997; 61: 862–867.
Bione S, D'Adamo P, Maestrini E, et al. A novel X-linked gene, G4.5, is responsible for Barth syndrome. Nat Genet 1996; 12: 385–389.
Codd MB, Sugrue DD, Gersh BJ, Melton LJ . Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy: a population-based study in Olmsted, County, Minnesota, 1975–1984. Circulation 1989; 80: 564–572.
Maron BJ, Gardin JM, Flack JM, et al. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation 1995; 92: 785–789.
Henry W, Gardin J, Ware J . Echocardiographic measurements in normal subjects from infancy to old age. Circulation 1980; 62: 1054–1061.
Vasan R, Larson M, Levy D, Evans J, Benjamin E . Distribution and categorization of echocardiographic measurements in relation to reference limits. The Framingham Heart Study: formulation of a height- and sex-specific classification and its prospective validation. Circulation 1997; 96: 1863–1873.
Hershberger RE, Ni H, Crispell KA . Familial dilated cardiomyopathy: echocardiographic diagnostic criteria for classification of family members as affected. J Card Fail 1999; 51: 203–212.
Kushner JD, Nauman D, Burgess D, et al. Clinical characteristics of 304 kindreds evaluated for familial dilated cardiomyopathy. J Card Fail 2006; 12: 422–429.
Baig MK, Goldman JH, Caforio AP, et al. Familial dilated cardiomyopathy: cardiac abnormalities are common in asymptomatic relatives and may represent early disease. J Am Coll Cardiol 1998; 31: 195–201.
Jerosch-Herold M, Sheridan DC, Kushner JD, et al. Cardiac magnetic resonance imaging of myocardial contrast uptake and blood flow in patients affected with idiopathic or familial dilated cardiomyopathy. Am J Physiol Heart Circ Physiol 2008; 295: H1234–H1242.
Hanson E, Hershberger RE . Genetic counseling and screening issues in familial dilated cardiomyopathy. J Genet Counseling 2001; 10: 397–415.
GeneReviews at GeneTests. Medical Genetics Information Resource. GeneTests/GeneClinics [cited, 2008 September 12, 2009]. Available at: http://www.genetests.org. Accessed August 16, 2010.
Morales A, Painter T, Li R, et al. Rare variant mutations in pregnancy-associated or peripartum cardiomyopathy. Circulation 2010; 121: 2176–2182.
Elkayam U, Akhter MW, Singh H, et al. Pregnancy-associated cardiomyopathy: clinical characteristics and a comparison between early and late presentation. Circulation 2005; 111: 2050–2055.
Demakis JG, Rahimtoola SH, Sutton GC, et al. Natural course of peripartum cardiomyopathy. Circulation 1971; 44: 1053–1061.
Hershberger RE, Cowan J, Morales A LMNA-related dilated cardiomyopathy. GeneTests/GeneClinics 2008, 2008. Available at: http://www.genetests.org. Accessed August 16, 2010.
Santorelli FM, Mak SC, El-Schahawi M, et al. Maternally inherited cardiomyopathy and hearing loss associated with a novel mutation in the mitochondrial tRNA(Lys) gene (G8363A). Am J Hum Genet 1996; 58: 933–939.
Li YY, Maisch B, Rose ML, Hengstenberg C . Point mutations in mitochondrial DNA of patients with dilated cardiomyopathy. J Mol Cell Cardiol 1997; 29: 2699–2709.
Arbustini E, Diegoli M, Fasani R, et al. Mitochondrial DNA mutations and mitochondrial abnormalities in dilated cardiomyopathy. Am J Pathol 1998; 153: 1501–1510.
Santorelli FM, Tanji K, Manta P, et al. Maternally inherited cardiomyopathy: an atypical presentation of the mtDNA 12S rRNA gene A1555G mutation. Am J Hum Genet 1999; 64: 295–300.
Marin-Garcia J, Goldenthal MJ, Ananthakrishnan R, Pierpont ME . The complete sequence of mtDNA genes in idiopathic dilated cardiomyopathy shows novel missense and tRNA mutations. J Card Fail 2000; 6: 321–329.
Mahjoub S, Sternberg D, Boussaada R, et al. A novel mitochondrial DNA tRNAIle (m. 4322dupC) mutation associated with idiopathic dilated cardiomyopathy. Diagn Mol Pathol 2007; 16: 238–242.
Michels VV, Moll PP, Miller FA, et al. The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N Engl J Med 1992; 326: 77–82.
McKenna C, Codd M, McCann H, Sugrue D . Idiopathic dilated cardiomyopathy: familial prevalence and HLA distribution. Heart 1997; 77: 549–552.
Grünig E, Tasman JA, Kücherer H, et al. Frequency and phenotypes of familial dilated cardiomyopathy. J Am Coll Cardiol 1998; 31: 186–194.
Mestroni L, Rocco C, Gregori D, et al. Familial dilated cardiomyopathy: evidence for genetic and phenotypic heterogeneity. Heart Muscle Disease Study Group. J Am Coll Cardiol 1999; 34: 181–190.
Morales A, Cowan J, Dagua J, Hershberger RE . Family history: an essential tool for cardiovascular genetic medicine. Congest Heart Fail 2008; 14: 37–45.
Hershberger R . Familial dilated cardiomyopathy. Prog Pediatr Cardiol 2005; 20: 161–168.
Richard P, Charron P, Carrier L, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 2003; 107: 2227–2232.
Van Driest SL, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ . Yield of genetic testing in hypertrophic cardiomyopathy. Mayo Clin Proc 2005; 80: 739–744.
Sen-Chowdhry S, Syrris P, McKenna WJ . Role of genetic analysis in the management of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol 2007; 50: 1813–1821.
McNally E, MacLeod H, Dellefave L Arrhythmogenic right ventricular dysplasia/cardiomyopathy, autosomal dominant. In: GeneReviews at GeneTests: Medical Genetics Information Resource (database online), 2008; initial posting, 2005. Available at: http://www.genetests.org.
Sen-Chowdhry S, Syrris P, Prasad SK, et al. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol 2008; 52: 2175–2187.
Sen-Chowdhry S, Morgan RD, Chambers JC, McKenna WJ . Arrhythmogenic cardiomyopathy: etiology, diagnosis, and treatment. Annu Rev Med 2010; 61: 233–253.
Hershberger R, Pinto J, Parks S, et al. Clinical and functional characterization of TNNT2 mutations identified in patients with dilated cardiomyopathy. Circ Cardiovasc Genet 2009; 2: 306–313.
Gupta P, Bilinska ZT, Sylvius N, et al. Genetic and ultrastructural studies in dilated cardiomyopathy patients: a large deletion in the lamin A/C gene is associated with cardiomyocyte nuclear envelope disruption. Basic Res Cardiol 2010; 105: 365–377.
Richard P, Villard E, Charron P, Isnard R . The genetic bases of cardiomyopathies. J Am Coll Cardiol 2006; 48( suppl A): A79–A89.
Bowles NE, Bowles KR, Towbin JA . The “final common pathway” hypothesis and inherited cardiovascular disease. The role of cytoskeletal proteins in dilated cardiomyopathy. Herz 2000; 25: 168–175.
Worman HJ, Courvalin JC . How do mutations in lamins A and C cause disease?. J Clin Invest 2004; 113: 349–351.
Menon S, Michels V, Pellikka P, et al. Cardiac troponin T mutation in familial cardiomyopathy with variable remodeling and restrictive physiology. Clin Genet 2008; 74: 445–454.
Hunt SA, Abraham WT, Chin MH, et al. ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: endorsed by the Heart Rhythm Society. Circulation 2005; 112: e154–e235.
Hunt SA, Abraham WT, Chin MH, et al. 2009 Focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults. A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed in Collaboration With the International Society for Heart and Lung Transplantation. J Am Coll Cardiol 2009; 53: e1–e90.
Heart Failure Society Of America Executive summary: HFSA 2006 Comprehensive Heart Failure Practice Guideline. J Card Fail 2006; 12: 10–38.
The SOLVD Investigators Effect of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fractions. N Engl J Med 1992; 327: 685–691.
Karkkainen S, Peuhkurinen K . Genetics of dilated cardiomyopathy. Ann Med 2007; 39: 91–107.
Rampersaud E, Kinnamon D, Hamilton K, et al. Common susceptibility variants examined for association with dilated cardiomyopathy. Ann Hum Genet 2010; 74: 110–116.
Badano JL, Katsanis N . Beyond Mendel: an evolving view of human genetic disease transmission. Nat Rev Genet 2002; 3: 779–789.
Bodmer W, Bonilla C . Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet 2008; 40: 695–701.
Ingles J, Doolan A, Chiu C, et al. Compound and double mutations in patients with hypertrophic cardiomyopathy: implications for genetic testing and counselling. J Med Genet 2005; 42: e59.
Girolami F, Ho CY, Semsarian C, et al. Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere protein gene mutations. J Am Coll Cardiol 2010; 55: 1444–1453.
Schwartz PJ, Priori SG, Napolitano C . How really rare are rare diseases?: the intriguing case of independent compound mutations in the long QT syndrome. J Cardiovasc Electrophysiol 2003; 14: 1120–1121.
Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC . Compound mutations: a common cause of severe long-QT syndrome. Circulation 2004; 109: 1834–1841.
Sen-Chowdhry S, Syrris P, Ward D, et al. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation 2007; 115: 1710–1720.
Kryukov GV, Pennacchio LA, Sunyaev SR . Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am J Hum Genet 2007; 80: 727–739.
Schork NJ, Murray SS, Frazer KA, Topol EJ . Common vs. rare allele hypotheses for complex diseases. Curr Opin Genet Dev 2009; 19: 212–219.
Ng SB, Turner EH, Robertson PD, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 2009; 461: 272–276.
Ng SB, Buckingham KJ, Lee C, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet 2010; 42: 30–35.
Cowan J, Li D, Gonzalez Quintana J, Morales A, Hershberger RE . Morphological analysis of 13 LMNA variants identified in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Circ Cardiovasc Genet 2010; 3: 6–14.
Acknowledgements
This work was supported by NIH award RO1-HL58626 (R.E.H.).
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Hershberger, R., Morales, A. & Siegfried, J. Clinical and genetic issues in dilated cardiomyopathy: A review for genetics professionals. Genet Med 12, 655–667 (2010). https://doi.org/10.1097/GIM.0b013e3181f2481f
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e3181f2481f
Keywords
This article is cited by
-
Transcriptome studies of inherited dilated cardiomyopathies
Mammalian Genome (2023)
-
Will “CLINICAL PROTEOMICS” lead to the discovery of new biomarkers for dilated cardiomyopathy (DCM)?
Journal of Proteins and Proteomics (2023)
-
Identification and functional characterization of BICD2 as a candidate disease gene in an consanguineous family with dilated cardiomyopathy
BMC Medical Genomics (2022)
-
DSP p.(Thr2104Glnfs*12) variant presents variably with early onset severe arrhythmias and left ventricular cardiomyopathy
BMC Medical Genetics (2020)
-
Patient experiences with hypertrophic cardiomyopathy: a conceptual model of symptoms and impacts on quality of life
Journal of Patient-Reported Outcomes (2020)
