
Abstract
Purpose: Israeli investigators have identified several relatively frequent disorders due to founder point mutations in Persian (Iranian) Jews, who, for nearly three centuries up to the Islamic Revolution of 1979, were completely isolated reproductively.
Methods: Using a community-based model previously employed with Tay-Sachs disease prevention, we developed a pilot program for the Persian Jewish community of greater Los Angeles. We screened for mutations responsible for four relatively frequent autosomal recessive conditions in Persian Jews in which effective interventions are available for each: Pseudocholinesterase deficiency (butyryl cholinesterase deficiency); Congenital hypoaldosteronism (corticosterone methyl oxidase II); Autoimmune polyendocrinopathy (autoimmune regulatory element); and Hereditary Inclusion Body myopathy.
Results: One thousand individuals volunteered. Mutations were assessed in saliva-derived DNA and were positive for 121/1000 butyryl cholinesterase deficiency; 92/1000 Hereditary Inclusion Body myopathy; 38/1000 corticosterone methyl oxidase II; and 37/1000 autoimmune regulatory element. Ten homozygous individuals (9 butyryl cholinesterase deficiency and 1 Hereditary Inclusion Body myopathy) and 10 “at-risk” couples (seven for butyryl cholinesterase deficiency and one each for the other three disorders) were identified. These frequencies are comparable with those in Israel and indicate an extraordinary level of inbreeding, as anticipated.
Conclusions: A carefully planned effort can be delivered to an “increased risk” community if detailed attention is given to planning and organization. However, availability of an effective intervention for those found to be “at-risk” or possibly affected, is essential before embarking.
Similar content being viewed by others

Main
In every isolated population, there are specific inherited disorders that tend to occur more frequently within that particular group than in the general population. This clustering of disorders is not unusual among close knit ethnic groups, because it reflects the common heritage and history of the individuals within the group. By studying such diseases within populations in which they are most common, it has been possible to identify the genes responsible for some of these disorders.1 Early disease detection and carrier screening programs rely on such data to identify at-risk individuals or couples, to implement intervention programs, and to inform other concerned family members that they are not at risk.
Jewish genetic disorders
In addition to having shared a religion, a cultural heritage, an oral tradition and a written language, Jews also share a common gene pool, dating back to their common origins more than 5700 years ago. Although the frequency of genetic diseases in general is no greater in Jews than in any other ethnic group, this shared genetic background has resulted in certain hereditary diseases occurring at a higher frequency in individuals of Jewish ancestry.2 Because of the historical migrations of Jews out of Israel over the millennia and the subsequent centuries long geographic separation of segments of the Jewish community, there are disorders that are more common among certain subgroups within the Jewish community, such as Ashkenazim, Sephardim and Persian Jews, and others.3 Some of these diseases, such as Tay-Sachs disease (TSD), Canavan disease, and familial dysautonomia, are relatively common among Jewish individuals of eastern and central European ancestry (Ashkenazim).4 Similarly, there are disorders that are more common among other branches of the Jewish people, such as familial Mediterranean fever and beta-thalassemia in Mediterranean/Iberian-derived (Sephardic) Jews and inclusion body myopathy in Persian/Iranian (Mizrahi) Jews.5
Research and education about some of the disorders clustered in Jewish families already have provided significant benefits to the Jewish community. Screening for carriers of TSD, for example, has virtually eliminated this devastating neurodegenerative disorder among Jewish infants.6,7 Research into the biochemical basis of Gaucher disease has lead to enzyme replacement therapy, which is of enormous benefit to affected individuals.8
The first community-based voluntary heterozygote screening program, launched in the early 1970s for the prevention of TSD in the Ashkenazi Jewish population (Jews of central and eastern European origins), was predicated on following three prerequisites.9
-
1
Nature of the disorder: severe, untreatable, and occurring predominantly in a defined population group.
-
2
Availability of an accurate, quality-controllable, relatively simple, and inexpensive heterozygote detection test (serum and leukocyte hexosaminidase A quantification).
-
3
An intervention acceptable to those found to be at risk (carrier: carrier couples) by which they could selectively have unaffected children of their own while preventing the birth of fetuses affected with TSD (through midtrimester amniocentesis or, subsequently, chorionic villous sampling “CVS” in the first trimester, with elective pregnancy termination when an affected fetus is identified).
Since inception in 1970, TSD “prevention” programs have been developed worldwide (North and South America, Israel, South Africa, Europe, and Australia). More than 1.5 million individuals have been tested voluntarily and more than 50,000 heterozygotes identified. Over 1500 couples have been found to be at risk, and 700 pregnancies in which the fetus was affected with TSD electively terminated (Kaback, unpublished data). Most importantly, more than 2800 healthy unaffected offspring have been born to these at-risk couples, many of which might never previously have been conceived in such families.10 In the United States and Canada, this has resulted in a more than 95% reduction in the prevalence of TSD in Jewish infants.6 This prototypic screening program also has served as a model which, with modification, has been implemented in Sardinia and other Mediterranean regions to greatly reduce the incidence of beta-thalassemia in those higher risk populations.11
The Persian Jewish screening program
Unlike the frequently migrating and emigrating Sephardic and Ashkenazi Jewish populations, the Persian (Iranian) Jewish population remained highly isolated and stable (and highly inbred) for >2400 years.12 The Persian Jewish community can trace its origins to the conquest of Palestine by King Nebuchadnezzar of Babylon in 586 BCE, and their subsequent Babylonian captivity. When Babylon was conquered by King Cyrus of Persia in 539 BCE, some of the then “freed-from-slavery” Jewish population returned to Palestine, but those who remained became the founders of a strong and unique Jewish community that has persisted to the present time.13 This group remained in Iran, completely isolated from other Jewish populations, until the Shah was deposed in 1979 with the Islamic Revolution. Within a few years of this event, the Jews of Iran left, almost in entirety, migrating in two major directions to North America and to Israel. The majority of those emigrating to the United States came to Southern California where it is now estimated that 50,000 to 75,000 Persian Jews reside. A significant, but smaller population, also now lives on Long Island, New York.
With the establishment of the State of Israel and the subsequent migration of Jews from many parts of the world to Israel, geneticists there began recognizing important distinctions, both in physical characteristics and in certain medical susceptibilities, between different Jewish groups.14–16 Because many of these distinct Jewish groups were highly inbred, it was not surprising that a number of different and distinct recessively transmitted hereditary disorders were identified more frequently, and in some instances almost exclusively, within one subgroup.
This certainly was the case with the Persian Jews, where a number of recessive disorders have been identified.5 With the advent of molecular diagnostic methods, it has become evident that a single, different, and unique “founder” mutation accounts for the underlying genetic etiology for nearly all the cited disorders in this population.14
Based on the findings of our Israeli colleagues, and the sizeable population of Persian Jews in Southern California, we queried whether similar frequencies of some of these mutant genes would be evident in our population. Also recognizing that several of the reported conditions met criteria similar to those previously described for the TSD program efforts (most particularly, the availability of very effective interventions for at-risk couples or affected individuals), we proceeded to develop a community-based education/voluntary testing/counseling program, modeled on the Tay-Sachs prototype, for the Persian Jewish community in Greater Los Angeles. We decided to initiate such an effort as a pilot project directed at education, counseling, and testing of 1000 individuals in this community. As this was to be an Institutional Review Board (IRB) approved research project, these first 1000 individuals were not to be charged for the tests performed.
We elected to focus our efforts on four conditions with increased frequencies in this population. These were selected primarily because of their reported relatively high frequencies in the Persian Jews migrating to Israel, the fact that three of the four disorders were associated with a single point mutation (and the fourth, with two mutant alleles), and lastly, and most critically, because these conditions were individually either entirely avoidable, treatable or, like TSD, preventable through reproductive counseling options. The four conditions selected were
Table 1 Persian Jewish genetic screening panel
-
1
Pseudocholinesterase deficiency E117,18: butyryl cholinesterase deficiency (OMIM #177400)—BCHE is the name for the genetic locus: an autosomal recessive condition common within the Persian and Iraqi Jewish populations. For purposes of clearer public education and understanding, we have called this condition “Anesthesia Sensitivity Syndrome.” Approximately 1 in 10 Persian Jews were found to be heterozygotes for a single point mutation in this gene (Table 1). Thus, 1 in 100 couples will both carry the mutant gene and each of their children will have a 25% risk of having two mutant genes and be affected with this disorder. The overall incidence among Persian Jews is therefore about 1 in 400 individuals. Most individuals do not know they are affected with pseudocholinesterase deficiency until they are exposed to specific drugs. When a person is affected with this disorder, certain anesthetic-related drugs (such as succinyl choline, a muscle relaxant) or certain local ester-based anesthetics (e.g., novocaine) cannot be broken down efficiently and therefore persist for a longer time in the body. These individuals may have prolonged effects of such agents, resulting in longer periods of difficulty in breathing or muscle weakness post procedurally. Complications can be avoided entirely by the selection, before surgery, of appropriate anesthetic and anesthesia-supportive drugs. Succinyl choline and certain ester-based local anesthetics (e.g., novocaine) should be avoided while other drugs not metabolized by BCHE can be used without complication.19
Table 1 Persian Jewish genetic screening panel -
2
Congenital hypoaldosteronism type 2: (OMIM #124080) CMOII deficiency is the designation given this disorder that results from a deficiency of activity of the enzyme, corticosterone methyl oxidase II.20 This cytochrome P450 isoenzyme is the product of the gene CYP11B2 located on chromosome 8q22. For the public, we have named this condition “Salt Losing Syndrome.” The disorder is manifested when an individual is homozygous for two point mutations at two allelic loci in this gene (Table 1).21 Heterozygosity is present when a person carries each of the two point mutations in a cis configuration. Approximately 1 in 30 Persian Jews are estimated to be heterozygous for this disorder and therefore 1 in 900 Persian Jewish couples are at risk of having a child with this condition. There is a wide range of manifestations in homozygotes affected with this salt losing disorder. In more severe forms, critical dehydration and shock may be apparent in the newborn period. If untreated, the infant may die. In less severe forms, individuals may have poor weight gain (failure to thrive) as a child, short stature, experience blood pressure irregularity, weakness, dizziness, or salt craving on into adulthood. When increased risk for this condition is identified before birth, the fetus or newborn can be tested and treatment begun early. A normal life span, growth, and development are expected for affected individuals with on-going early, simple, and inexpensive mineralocorticoid replacement therapy.
-
3
Autoimmune polyendocrinopathy syndrome, type 122,23 (OMIM #240300) is an autosomal recessive condition. We have called this condition “Multiple Hormone Deficiency” for easier understanding. The gene involved in this disorder is AIRE, for autoimmune regulatory element, and its role is thought to relate to early “self” recognition in the fetal thymus. The point mutation (Table 1) is present in approximately 1 in 50 Persian Jews (heterozygotes) and 1 in 2500 Persian Jewish couples are at risk of having a child affected with this condition. Depending on which glands are affected, this condition can be characterized by many symptoms, including any of the following: skin infections (particularly Candidiasis), fatigue, muscle weakness, loss of appetite, nausea, vomiting, tightness/tingling of the lip or fingers, twitching and painful spasms in the muscles, dehydration, anxiety/depression, hair loss, and infertility, depending on the particular hormonal deficiencies present. Affected individuals usually do not experience all these symptoms. When diagnosed, this disorder is readily treated with hormone replacement therapy depending on which hormonal symptoms are evident.
-
4
Hereditary inclusion body myopathy (HIBM) is transmitted as autosomal recessive condition24,25 (OMIM #600737). For public purposes, we call this condition “Hereditary Muscle Disorder.” A single point mutation in the gene GNE is found in approximately 1 in 20 Persian Jews, and 1 in 400 Persian Jewish couples are therefore at risk of having a child affected with this condition. HIBM is characterized by progressive muscle weakness of the arms and legs. The symptoms usually appear in the 20th to 30th year of life with difficulty walking. The progression of the disease is gradual but most affected individuals eventually become severely debilitated within a decade after onset and lose their ability to walk. Preconception genetic counseling or prenatal diagnosis in early pregnancy is available to at-risk couples. The fundamental defect in this condition remains unclear, but like other dystrophies, considerable research directed to therapy is ongoing.
Each of these conditions is of sufficient morbidity and frequency to warrant efforts either to identify affected individuals before symptoms are evident (where effective treatment is available), or, where no treatment is at hand, to be able to identify individuals or couples at high risk for disease in their offspring and to provide them with counseling and assistance with their reproductive decisions.
MATERIALS AND METHODS
Program development
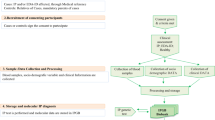
To consider the pilot effort, we needed first to be certain whether we could identify the five mutations associated with these disorders, in as simple, inexpensive, and totally accurate a fashion as possible. Also, the methods had to be capable of high throughput application as sizeable numbers of samples would be expected, with rapid turnover needed to report results in a reasonable time frame.
Stored blood and DNA samples were recovered anonymously from approximately 200 Persian Jewish individuals who had been previously tested in community testings offered for identifying heterozygotes of HIBM or TSD. Using the DNA from these samples, with the methods described, it was clear that these mutations were detectable and abundant in our Southern California Persian Jewish community.26 Next, to facilitate community-based testing and minimize costs, we established that saliva-derived DNA samples were readily usable for these determinations and blood testing would not be necessary. Using saliva, and not requiring a blood sample, was expected to greatly improve compliance among community members. A 2–5 mL saliva sample is easily obtained (compared with venapuncture with its frequently perceived unpleasantries) and 1–10 μg of DNA readily derived using the Oragene Saliva Collection Kit™ (Kanata, Ontario, Canada).
As with the earlier TSD programs, a series of steps were then undertaken to optimize the success of the program:
-
Phase I. Before embarking, it was clear that additional funds would be necessary to perform such an effort: laboratory equipment and supplies, printing (brochures and flyers, posters, etc.), testing supplies, counseling, and other additional office and laboratory personnel. Cedars-Sinai Medical Center agreed to meet the initial costs of establishing the laboratory and supply aspects of the program. The Jewish Community Foundation of Los Angeles responded positively to our grant application for 3 years of support for the genetic counselor/coordinator needed to implement the program.
-
Phase II. Several community leaders, including physicians, organizational leadership, and rabbis serving this population were consulted to assess their responses and ideas about such an effort in their community. With their uniformly enthusiastic support for the idea and their willingness to help and participate, we continued to proceed. From these leaders, an advisory committee of 25–30 members was formed for ongoing input and feedback in further planning, and for continued input after the program began. Possible individual, as well as community wide, stigmatization was thoroughly explored with this committee and was not felt to be a major issue with the educational, counseling, and testing program we outlined.
-
Phase III. Counselor/Coordinator recruitment and IRB approval were obtained. Public relations planning was initiated. A 12-minute video DVD was produced, presenting the background, objectives, and nature of the program. Other educational materials were produced: flyers, brochures, power point presentations, internet sites (U-Tube, Facebook, etc.), and a web site.
-
Phase IV. After all the above were accomplished, directed educational sessions were held (special lectures, grand round presentations for OB/GYN, Internal Medicine, Pediatrics, and Family Medicine), community organizational meetings, sermons, and invited presentations at synagogues after weekly services or at special events. These efforts were critically timed so that the educational phase occurred only in the 4 or 5 weeks before the community-based testing sessions.
In addition, extensive efforts were made to enhance public education through community and citywide media coverage: newspaper and television presentations again were timed, where possible, to immediately precede the community opportunities. For each testing, a special “training” session was held for 15–20 volunteers who staffed each of the testing sites.
Organization of testings
Six testing opportunities were made available to the community between July and October of 2009. These were held on either Sunday afternoons (2–5 PM) or weekday evenings (5–8 PM) to facilitate community participation. Three sessions were held at the two synagogues where the majority of Persian Jews in Los Angeles worship. One was held at a community center in the San Fernando Valley, a part of Los Angeles County, and two were held as evening sessions at Cedars-Sinai Medical Center. Participants were confined to persons ≥16 years.
For each session, to control numbers and to be able to appropriately staff the testing, individuals made phone reservations and were assigned appointments at 1-hour shifts through the 3-hour testing session. Genetic counselors volunteered at each testing and provided information, answered questions, and assured required IRB completion of all informed consent and HIPAA documents. Each individual completed a questionnaire that queried national and Iranian city-of-origin information, as well as whether they had blood relatives with any of the four disorders under consideration. Each completed a self-addressed envelope to themselves in which most test result letters would be sent (if negative for all mutations). Bar code stickers on all tubes and paperwork sheets made samples and information essentially anonymous to everyone except selected program staff (the genetic counselor only in nearly all cases). A 2 to 5-mL saliva sample was collected from each person in an Oragene saliva Collection Kit™ (Kanata, Ontario, Canada), bar-coded, mixed as directed in the collection vessel, and then maintained at room temperature until delivery to the laboratory for subsequent analysis.
Laboratory methods
DNA was isolated from the stabilized saliva using a 96-well Qiagen (Valencia, CA). Purified DNA was quantified using a Nanodrop-1000 spectrophotometer and diluted to 5 ng/mL. Saliva samples of 2–5 mL were obtained from each participant and 1–10 μg of DNA extracted in all cases so that no individuals were recalled for further sampling. Genotyping for AIRE, BCHE, and GNE were performed using a Taqman allele specific probes real-time PCR and detected on an ABI SDS 7500 system. Interpretable results were obtained in all 1000 samples. A common pair of primers was used for each reaction with allelic discrimination by allele specific probes labeled with fluorophores FAM or VIC. Samples were genotyped using the allelic discrimination analysis from Applied Biosystems with results confirmed by real-time amplification curve review.
Genotyping for the CYP11B2 R181W was performed using a fast cycling real-time PCR with fluorescence resonance energy transfer hybridization probes determining the genotype by melting curve analysis on the Roche LightCycle™. Custom made primers were designed to selectively amplify the CYP11B2 region. Allelic discrimination is accomplished by use of hybridization detection probes that bind to the region of the 181 codon regardless of genotype but are a perfect match to the mutant allele. Samples yield a single peak at ∼52.5°C if homozygous wild-type, a single peak at 60.5°C if homozygous mutant and peaks at both temperatures if heterozygous.
Because of the highly conserved nature of the CYP gene, genotyping of the V386A and R181W were unable to be performed using standard real-time hydrolysis probes under conserved conditions. The CYP11B2 V386A is performed using an unlabeled probe detected by high-resolution melting curve analysis. The samples are amplified in an asymmetric PCR reaction with a 10:1 forward:reverse primer concentration. The reaction mixture includes a C3 blocked and dephosphorylated probe which in the presence of LC fluorophores™, Idaho Technologies green+, a fully saturating base intercalator, discriminated between the alleles by the melting temperature of each specific allele. Melting curve data are collected using the LightScanner (Idaho Technologies, Salt Lake City, UT).
Communication of results
By bar code identification number, laboratory results were transmitted electronically to the Program Genetic Counselor who was responsible for most result transmissions as follows:
-
a
Noncarrier of each of the four traits: a form letter stating this finding was sent in the self-addressed envelope to that individual.
-
b
Heterozygote for 1 (or more) mutations: before mailing any results to all participants of each particular testing, each person found to be positive for any of the four conditions was contacted by a phone call from the genetic counselor. The finding was discussed, questions answered, and the individual was invited to the center for further counseling if they chose. Also explained was the importance of this finding for their close “blood” relatives. They were informed that we will provide them (the carrier) with letters and brochures for them to give or send to their relatives explaining this finding and providing information about the disorder(s) involved, and instructions for how they, if they choose, can be tested. This, and their own result letter, was then sent to them in their own self-addressed envelope.
-
c
Homozygotes: it was anticipated from the outset of the Program, that with the relatively high mutation frequencies for these disorders in this population, homozygous (affected) individuals could be identified. This possibility was also likely in light of the considerable variability in phenotype and symptoms in homozygotes with both the polyendocrine disorder and the hypoaldosterone condition and also the late onset of symptoms in many individuals afflicted with HIBM. Further, the absence of phenotype in psuedocholinesterase deficient individuals, unless exposed to specific inciting pharmaceuticals, or their unawareness of the condition even after having been exposed, makes this expectedly frequent homozygous genotype quite likely to be first identified by our program.
For the polyendocrine and aldosterone deficiency conditions, presymptomatic detection of homozygotes was optimal because these are readily treatable conditions should symptoms arise or subtle ones already be evident but not appreciated. With psuedocholinestersase homozygosity, this is a “noncondition” if specific drugs are avoided with general or local anesthesia. HIBM, however, becomes much more problematic, because eventual progressive debilitation will occur. Such a finding could be highly devastating to a young adult who sees him or herself as a completely “healthy” person.
For these reasons, all individuals found to be homozygous for any of the disorders were contacted first by phone by a physician-geneticist or counselor and requested to come in to meet with the physician geneticist. This allows not only for oral and written information to be communicated directly but also permits appropriate physical examination, laboratory, and/or radiologic studies, or referral where appropriate.
d. At-risk couples: because each of the conditions screened for are transmitted with an autosomal recessive pattern of inheritance, couples at risk for affected offspring (heterozygote/heterozygote, or heterozygote/homozygote couples) are primary goals for identification. Again, because of the need for more expansive counseling, each couple so identified was called and asked to come in to the Center for genetic counseling and to receive written materials as described previously.
RESULTS
With 10–15 community volunteers, 3–6 genetic counselors, and 4–6 saliva collecting stations, participants moved smoothly through each of the testings in a timely fashion. Between 150 and 250 persons were tested at each of the six community-based testings. On a few occasions, children <16 years of age were brought by their parents to the testing, but generally were not tested. Video presentations of program principles and goals were repeatedly presented at each site, and participants were encouraged to observe the 12-minute video and review the factual brochure before proceeding.
Exactly 1000 persons were tested in the pilot phase of the program. Results are shown in Table 2. A total of 288 individuals were identified as heterozygotes for at least one of the four disorders involved. Ten persons were found to be homozygotes (9 for the BCHE mutation and 1 for the mutation underlying HIBM). Further, 10 “at-risk” couples were discovered (1 for AIRE, 7 for BCHE, 1 for HIBM, and 1 for CMOII). These are extraordinary results and clearly support the likelihood that a very high degree of inbreeding exists among these respondents. Multiple members of the same families, often with different last names, frequently volunteered for testing. Because there may have been a strong selection bias among those who chose to come for testing because of known affected family members, whose names we could not ascertain for confidentiality reasons, it is not possible to cite exact frequencies of these mutations in the Los Angeles Persian Jewish population. However, it is quite evident that each of these gene mutations is abundant in the Persian Jews tested here and comparable to frequencies reported in Israel.27 Indeed, 29 persons were found to be heterozygotes for two of the four disorders, another individual was homozygous for pseudocholinesterase deficiency and heterozygous for two of the other conditions, and two other persons were identified as heterozygotes for three of the four disorders!
Homozygotes
Of particular note was the finding of 10 homozygous individuals, 9 for psuedocholinesterase deficiency, and 1 for HIBM. When contacted, several of the BCHE homozygotes had histories of previous symptoms associated with anesthesia (particularly spinal or epidural with novocaine). None had been diagnosed with this disorder. The individual homozygous for the HIBM mutation (age 27) was completely asymptomatic. This young woman has been carefully counseled by one of us (D.L.R.) and subsequently referred to a neuromuscular specialist. There, her examination, with extensive muscle testing included, was within normal limits and she is now being followed every 6 months.
The nine individuals found to be homozygous for the pseudocholinesterase mutation were counseled about their susceptibility and provided with a list of agents, which they should warn physicians or other health professionals if local anesthetic agents like novocaine or general anesthesia-associated agents like succinyl choline are being considered in their care. In both instances, other agents can be employed without complications. Medic-alertTM bracelets were recommended.
At-risk couples
Ten couples were identified in which both spouses were heterozygotes for the same mutation and therefore at risk for the respective disorder in their offspring. In cases where couples were detected to be at risk, testing was offered to all of their offspring. In the case of the hypoaldosterone disorder, both members of the identified at-risk couple were found to be heterozygous at both alleles associated with this condition. Each of the psuedocholinesterase at-risk families were informed of the avoidability of associated problems and advised to have all children tested and, if found to be homozygous, to understand the importance of informing any physician of this before any local or general anesthesia-associated agents are administered. It was also suggested that homozygous individuals wear a Med Alert™ bracelet warning health professionals of this vulnerability.
Families at risk for the polyendocrine deficiency as well as those at risk for the hypoaldosterone disorder were counseled as to the relative facility and minimal expense of treatment for each of these conditions. Here testing shortly after birth is recommended and appropriate treatment initiated if and when it becomes indicated. Prenatal testing by amniocentesis or CVS for these two conditions seems less applicable if diagnosis and treatment, if appropriate, are initiated when necessary. Such testing could be used, however, to forewarn the family and pediatrician of the homozygous condition of the fetus to be alerted and prepare for possible exigencies after birth. As pseudocholinesterase deficiency is clinically insignificant if specific drugs are avoided by homozygotes, it is recommended to these couples that their newborns be tested and, if affected, that they wear the bracelet and be fully informed at an appropriate age.
With couples found to be at risk for HIBM, counseling alternatives like those associated with TSD counseling are offered. However, because of the late-onset and mild symptoms in some patients, this becomes even more difficult than counseling the family at risk for classical infantile TSD. One important new option, however, which lessens somewhat the burden for couples at risk is the alternative provided by preimplantation diagnosis after in vitro fertilization. This option, although complex and expensive, provides a means by which only unaffected pregnancies can result.
CONCLUSION
We have demonstrated that a carefully planned effort of community education, genetic testing, and follow-up counseling can be delivered effectively to a targeted “increased risk” community if detailed attention is given to the planning and organizational aspects of such a program. Clearly, the availability of realistic and effective interventions for those found to be affected or at risk for disease greatly facilitates such efforts. It should be pointed out, however, that the initial phase of reporting results to community members only initiates the second, perhaps more challenging aspect of the program, e.g., contacting and providing services to first and second degree relatives of each of the identified hetero and homozygotes discovered. Each creates its own “snow-ball effect,” with demands for communication, counseling, and testing to potentially a large number of widely dispersed relatives. Further, this pilot testing was offered to the population at no cost, removing any financial barrier that might otherwise inhibit community members' compliance. It remains to be seen if insurance offsets, local philanthropy, or individual payment for testing costs will be possible in the future to maintain similar enthusiastic responses from this community. Financial restraints also may perturb the testing of relatives as well, since the “no-cost phase” of the program has ended and their testing will require payment. We hope these fiscal obstacles can be overcome, because it is certainly our intention to continue these efforts in this area.
As with the experience with TSD prevention, focused and carefully planned efforts can be used to mobilize a community with little prior knowledge of genetic aspects of certain diseases. This model potentially has application to many other “ethnic” disorders but with one major caveat. Merely knowing the specific genetic defect underlying a given condition in a defined subpopulation, and having the ability in the laboratory to identify the mutation, the deficiency or the disease predisposition—while necessary to perform targeted community-based testing—is not in itself, sufficient to do so. It is the availability of an acceptable intervention for those found to be positive through testing that justifies mounting such an effort. Also, there is a critical need for input from organizational, religious, and medical leadership of the targeted community before embarking on such a venture. The dynamics of each community may be vastly different making genetic testing efforts such as that described here more or less problematic. Accordingly, careful advanced planning and organization are critical to the success of such an effort.
References
Emery AE, Rimoin DL . Emery and Rimoin's principles and practice of medical genetics, 5th ed. New York, NY, Churchill Livingstone, 2006.
Goodman RM . Genetic disorders among the Jewish People. Baltimore, MD, Johns Hopkins University Press, 1979.
Oster H . A genetic profile of contemporary Jewish populations. Nat Rev Genet 2001; 2: 891–898.
Zlotogora J . Mendelian disorders among Jews. Jerusalem, Israel, Haddasah University Press, 1999.
Zlotogora J . Hereditary disorders among Iranian Jews. Am J Med Genet 1995; 58: 32–37.
Kaback MM . Population-based genetic screening for reproductive counseling; the Tay-Sachs disease model. Eur J Pediatr 2000; 159: 192–195.
Kaback MM, Lim-Steele J, Dabholkar D, Brown D, Levy N, Zeiger K . Tay-Sachs disease—carrier screening, prenatal diagnosis and the molecular era. An international perspective, 1970 to 1993. The International TSD Data Collection Network. JAMA 1993; 270: 2307–2315.
Weinreg NJ, Charrow J, Andersson HC, et al. Effectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher Registry. Am J Med 2002; 113: 112–119.
Kaback MM, Zeiger RS Heterozygote detection in Tay-Sachs disease: a prototype community screening program for the prevention of recessive genetic disorders. In: Volk BW, Aronson SM (eds) Advances in experimental medicine and biology. New York, NY, Plenum Press, 1972.
Aronson S Epidemiology. In: Volk BW (ed) Tay-Sachs disease. New York, NY, Grune & Stratton, 1964.
Cao A, Galanello R . Beta-thalassemia. Genet Med 2010; 12: 61–67.
McBrewster J, Miller FP, Vandome AF . Persian Jews: history of Jews in Iran. Mauritius, Alphascript Publishing,
Zohar Z . Sephardic and Mizrahi Jewry: from the Golden Age of Spain to Modern Times. New York, NY, NYU Press, 2005.
Israeli Ministry of Health's Golden Helix database. Available at: http://www.health.gov.il/Download/pages/tablejews012010.pdf. Accessed May 1, 2010.
Zlotogora JV, Bach G, Munnich A . Molecular basis of Mendelian disorders among Jews. Mol Genet Metab 2000; 69: 169–180.
Zlotogora J, van Baal S, Patrinos GP . Documentation on inherited disorders and mutation frequency in the different religious communities in Israel in the Israeli National Genetic Database. Hum Mutat 2001; 28: 944–949.
Szeinberg A, Pipano S, Assa M, Medalie J, Neufeld H . High frequency of atypical pseudocholinesterase gene among Iraqui and Iranian Jews. Clin Genet 1972; 3: 123–127.
Neville L, Gnatt A, Padan R, Seidman S, Soreq H . Anionic site interactions in human butyrylcholinesterase disrupted by two adjacent single point mutations. J Biol Chem 1990; 265: 20735–20738.
Alexander D Pseudocholinesterase deficiency. eMedicine. 2009. Available at: http://emedicine.medscape.com/article/247019-overview. Accessed March 12, 2010.
Cohen T, Rosler T . Selective hypoaldosteronism in Iranian Jews: an autosomal recessive trait. Clin Genet 1977; 11: 25–30.
Pascoe L, Curnow K, Slutsker L, Rosler A, White P . Mutations in the human CYP11B2 (aldosterone synthetase) gene causing corticosterone methyloxydase II deficiency. Proc Natl Acad Sci U S A 1992; 89: 4996–5000.
Zlotogora J, Shapiro M . Polyglandular autoimmune syndrome in Iranian Jews. J Med Genet 1992; 29: 834–836.
Bjorses P, Aaltonen J, Vikman A, et al. Genetic homogeneity of autoimmune polyglandular disease type I. Am J Hum Genet 1996; 59: 879–886.
Argov Z, Yaron R . “Rimmed vacuole myopathy” sparing the quadriceps. A unique disorder in Iranian Jews. J Neurol Sci 1984; 64: 33–43.
Mitrani-Rosenbaum S, Argov Z, Blumenfeld A, Seidman C, Seidman J . Hereditary inclusion body myopathy maps to chromosome 9p1–q1. Hum Mol Genet 1996; 5: 159–163.
Riley-Portuges CA, Lopategui JR, Kaback MM, Rimoin DL Development of a Rapid Genetic Screening Panel for the Southern Californian Persian Jewish Community. Proceedings of Annual Meeting American Society of Human Genetics 2009.
Zlotogora J, van Baal S, Patrinos GP . The Israeli National Genetic Database. Isr Med Assoc J 2009; 11: 373–375.
Acknowledgements
Supported by Jewish Community Foundation of Los Angeles.
The authors thank Drs. Issy Lerer, PhD, Esther Leshinsky-Silver, Daniel Darvish, Joyce Lim-Steele, and Deepti Dabholkar for providing sample mutant DNA and Zvi Borochovitz, Motti Shohat, Joel Zlotogora, and Elon Pras for their invaluable advice, Sue Lief for her hard work in organizing the screenings, Nancy Kramer, Tara Funari, Raluca Kerz, Tali Geva, Eyal Reinstein, Angela Sun, Krishna Singh, Grace Noh and Iris Mencias for providing genetic counseling at the screenings, Arleen Hernandez, Chantel Manchester, Sheryl-Lyn Reyes and to the many other community volunteers who assisted us.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Kaback, M., Lopatequi, J., Portuges, A. et al. Genetic screening in the Persian Jewish community: A pilot study. Genet Med 12, 628–633 (2010). https://doi.org/10.1097/GIM.0b013e3181edef5b
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e3181edef5b
Keywords
This article is cited by
-
Consanguineous marriages
Journal of Community Genetics (2012)
