
Abstract
Purpose: To define the prevalence of PTEN mutations in a clinical cohort of pediatric subjects with autism spectrum disorders (ASDs), developmental delay/mental retardation (DD/MR), and/or macrocephaly and to assess genotype–phenotype correlations.
Methods: Medical records of patients who had clinical PTEN gene sequencing ordered through our institution between January 1, 2005 and December 31, 2007 were abstracted to confirm genetic test results and medical diagnoses. Phenotypic information related to the diagnoses, prenatal history, early developmental milestones, physical characteristics, and family history for those with a confirmed PTEN mutation was also recorded.
Results: One hundred fourteen patients were tested during this time period for indications of ASDs (N = 60), DD/MR (N = 49), or macrocephaly only (N = 5). Eleven mutations were identified: five in patients with ASDs and six in those with DD/MR, resulting in a prevalence of 8.3% and 12.2% in these respective clinical populations. All individuals with a PTEN mutation had significant macrocephaly (>2.0 SD)
Conclusions: These data illustrate that PTEN gene sequencing has a high diagnostic yield when performed in a selected population of individuals with ASDs or DD/MR and macrocephaly. Germline mutations in PTEN are an important, identifiable etiology among these patients.
Similar content being viewed by others

Main
The phosphatase and tensin homologue deleted on chromosome 10 (PTEN) gene (MIM 601728) is a tumor suppressor gene that functions as a dual-specificity phosphatase active in numerous pathways involved with cellular growth.1,2 It functions primarily as a lipid phosphatase acting to down-regulate the phosphoinositol 3-kinase/AKT pathway. This pathway is involved in cell proliferation, and its dysregulation has been associated with overgrowth syndromes.3 Heterozygous germline mutations in PTEN have been identified in a family of related disorders, including Cowden syndrome (CS—MIM 158350), Bannayan-Riley-Ruvalcaba syndrome (BRRS—MIM 153480), Proteus and Proteus-like syndromes. Individuals with CS typically develop multiple benign hamartomas and have an increased risk of certain cancers, particularly of the breast, uterus, and thyroid. BRRS is characterized by macrocephaly, hamartomas (including lipomas, hemangiomas, or intestinal polyps), penile freckling in males, and often developmental delay with childhood onset. Approximately 80% of individuals who meet diagnostic criteria for CS and 60% who meet clinical criteria for BRRS have detectable PTEN mutations.4 Although genotype–phenotype correlations have been sought, it seems that CS and BRRS may actually be one condition with variable expressivity and presentation at different ages.5 Because of phenotypic overlap among these disorders, and with Proteus-like syndromes, they have collectively been termed the PTEN hamartoma tumor syndromes.6
Although PTEN may be most recognized for its role in carcinogenesis, several recent reports have identified heterozygous germline PTEN mutations in children with autism spectrum disorders (ASDs) and macrocephaly where a parent had previously been diagnosed with CS.7–9 On the basis of these findings, Butler et al.10 screened 17 children with an ASD and macrocephaly with no family history of CS and identified PTEN mutations in three of them. Subsequently, we reported additional PTEN mutations in two unrelated children with significant macrocephaly and autism.11 Finally, Buxbaum et al.12 screened 88 patients with an ASD and macrocephaly and discovered a de novo missense mutation (D326N) in one subject.
Although these findings have led the authors to conclude that PTEN sequencing is warranted in patients with ASDs and significant macrocephaly, the prevalence of pathogenic PTEN mutations within this population remains unknown. We sought to define the detection rate of PTEN mutations in a cohort of pediatric patients undergoing clinical gene sequencing between January 1, 2005 and December 31, 2007. Concurrently, we performed a retrospective chart review to confirm the diagnosis of autism, significant developmental delay/mental retardation (DD/MR), and macrocephaly, and recorded the PTEN mutation status. Phenotypic information related to the prenatal history, early developmental milestones, diagnosis of ASD, physical characteristics, and family history for those with and without PTEN mutations was also recorded, with a goal of further delineating possible genotype–phenotype correlations.
MATERIALS AND METHODS
After institutional review board approval, we obtained a list of patients who were tested clinically for PTEN mutations through our institution between January 1, 2005 and December 31, 2007. We developed a guideline for genetic testing of children with ASD at our hospital in January 2005, which included PTEN testing for those individuals with ASD and macrocephaly.13 This guideline is used in virtually all children with ASD presenting to our institution.
ICD-9 diagnosis codes and testing information were obtained for each patient. Clinical records were requested from ordering providers, most commonly from the Divisions of Molecular and Human Genetics and Neurology and the Developmental Disabilities/Autism Clinics at Nationwide Children's Hospital. Records were abstracted by two board-certified genetic counselors (E.A.V. and M.P.). Prenatal history, current and past medical diagnoses; findings on physical exam, such as weight, height, and occipital-frontal circumference (OFC); criteria for autism diagnoses (if applicable); family history including cancers, psychiatric diseases, and/or ASDs; and indication for testing and genetic test results were recorded. Information abstracted from medical records was collected and stored in a password-protected Microsoft Access database. OFC Z scores (by age and sex) were calculated using the Abase Anthropometry Database version 1.2,14 which is based on Centers for Disease Control growth charts for ages 0–2 years and Canadian normative data for ages 3–18 years.15,16 For the purpose of data analysis, subjects were categorized as having an ASD, DD/MR, and/or macrocephaly. Patients who did not have a diagnosis of ASD, DD/MR, or macrocephaly, and those who were subsequently found to have an alternate genetic diagnosis to explain their findings, were excluded from data analysis.
All PTEN gene sequencing was performed by the Clinical Laboratory Improvement Amendments (CLIA) certified Molecular Pathology Laboratory at The Ohio State University. Both strands of the PTEN coding region and the intron–exon junctions were sequenced. The two strands of the polymerase chain reaction (PCR) products were sequenced with the BigDye terminator cycle sequencing kit v1 (Applied Biosystems, Foster City, CA) and analyzed on capillary electrophoresis on an ABI Prism 3100 sequencer (Applied Biosystems). Alterations from the wild-type sequence were identified using the software program Mutation Surveyor (SoftGenetics, State College, PA). The PTEN promoter was not examined. Sequence data on 800 normal control alleles has been previously collated by this clinical laboratory.
We searched the literature and the Human Mutation Database for the PTEN gene (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=PTEN) to determine if the mutations identified in our subjects had been previously described. Novel mutations were analyzed in POLYPHEN17 (http://genetics.bwh.harvard.edu/pph/) to determine possible pathogenic significance. Cross species comparison for sequence conservation was performed by CLUSTALW alignment of protein sequences downloaded from the Ensembl website database (http://www.ensembl.org/index.html—March 2008, release 49).
One subject (Patient 5) was studied in more detail for possible splice alterations in PTEN. Primary white blood cells and Epstein-Barr virus–derived lymphoblastoid cell lines were used for analysis in the subject. Lymphoblastoid cell lines for both parents and one normal control were also used. Total RNA was obtained with the RNeasy Mini Kit (Qiagen, Valencia, CA). Genomic DNA was removed by DNase, and cDNA was prepared using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Reverse transcriptase PCR products covering from exons 4 to 9 were amplified with the previously described primers 4F and 9R.17
We used Stata version 10 (Stata Corp, College Station, TX) for all descriptive and comparative statistical analysis. Categorical variables were compared for differences by χ2 or Fisher exact test. For χ2 analysis, one degree of freedom was used. Quantitative variables were first assessed to ensure the group variances were equal by the Bartlett test of homogeneity, and then analyzed by t test. Significance was set at P < 0.05.
RESULTS
Subject characteristics
PTEN gene sequencing for germline mutations was performed on 136 subjects in our Molecular Pathology Laboratory during the 3-year period analyzed (see Methods section). Eight of these patients did not have a diagnosis of macrocephaly, DD/MR, or an ASD and were excluded from data analysis. Genetic testing revealed an alternate genetic diagnosis in another 12 patients: two patients with 46,XY,dup(15)(q11.2q13); and one patient each with: mitochondrial complex II deficiency; duplication of MeCP2; 45X,dic(Y;22)(q12:q12:p11.2); de novo del 9q22.32; de novo del 22q11.2; dup(22)(q11.2)mat; del(1)(q21.1)mat; del(3)(p26.3)mat; del(X)(p22.22)mat; and dup(13)(q32.3q33.1)mat. Finally, two subjects tested were first-degree relatives of a proband; one subject was tested after a PTEN mutation was identified in his identical twin and another was tested at the same time as her mother who had DD/MR and macrocephaly. This resulted in 22 individuals being excluded from our sample, leaving 114 for analysis.
Characteristics of the 114 subjects are summarized in Table 1. Patients ranged in age from 3 months to 35 years (mean 5.8 years; median 4.3 years). Eighty-five were male and 29 were female (ratio 3.4:1); this ratio is reflective of the increased prevalence of ASDs in males.18 A majority of the subjects were white (82%).
Sixty subjects had a diagnosis of an ASD based on DSM-IV criteria. Twenty-five of the subjects were diagnosed with an ASD by a developmental pediatrician, eight were diagnosed through a multidisciplinary evaluation (including a psychologist and developmental pediatrician at a minimum), three by a neurologist, and two by a psychiatrist, with the remaining being diagnosed by other physicians. The diagnosis of an ASD was confirmed in 13 subjects using the Autism Diagnostic Observation Scale.19 As depicted in Figure 1, 30 of 60 (50%) subjects with ASD also had DD/MR. Twenty-seven (45%) of subjects with ASD also had macrocephaly. Forty-nine subjects without ASD had a diagnosis of DD (age < 5 years) or MR (age > 5 years). Of these, 32 (65%) were macrocephalic. The remaining five subjects in our cohort were evaluated for an indication of macrocephaly without ASD, DD, or MR.

Diagnoses of study population: Sixteen subjects had an autism spectrum disorder (ASD) without developmental delay/mental retardation (DD/MR) or an occipital-frontal circumference (OFC) > 2.0 (macro). Seventeen subjects had DD/MR only. Seventeen subjects had ASD and DD/MR without macro. Fourteen subjects had ASD and macro, two of whom were found to have a PTEN mutation (PTEN). Thirty-two subjects had DD/MR and macro, six of whom were found to have PTEN. Thirteen subjects had ASD, DD/MR, and macrocephaly, three of whom were found to have PTEN. Five subjects had macrocephaly only; none of these subjects were found to have PTEN.
PTEN gene sequencing was most commonly ordered by clinical geneticists (N = 67), whereas developmental and general pediatricians performed testing in 35 cases. In addition to PTEN sequencing, additional diagnostic testing was performed on most of the subjects. Peripheral blood chromosomal analysis (at least 550 band metaphase spread) was performed in 97 subjects; DNA testing for fragile X syndrome was obtained in 88; and targeted BAC microarray (including Spectral 2600 and Constitutional, Signature V.3.0 and V4.0 and Baylor College of Medicine's chromosomal microarray) was completed in 73 subjects. Two subjects had abnormalities identified on chromosome analysis that were present in one parent, one of which is a known chromosomal variant. Two had abnormalities on microarray that were also present in one of the parents and are known copy number variants in the general population (as determined by accessing the Database of Genomic Variants, http://projects.tcag.ca/variation/) and are thus considered normal variants.
Prevalence and description of PTEN mutations in our subject population
Eleven PTEN mutations were identified in our clinical cohort, resulting in a prevalence of 9.6%. Clinical summaries for Patients 1 and 5 have been reported previously.11 Clinical case reports for the other nine patients are provided in the online Supplementary material with relevant data (see Supplemental Digital Content 1, http://links.lww.com/A687), including mutation descriptions, summarized in Tables 2 and 3.
Five subjects with a DSM-IV diagnosis of an ASD had a PTEN mutation, resulting in a prevalence of 5 of 60 (8.3%) in this subgroup (Table 2). Six PTEN-positive subjects had a diagnosis of DD and macrocephaly without ASD resulting in a prevalence of 6 of 49 (12.2%) in the DD/MR subgroup.
The distribution of OFC Z scores in our cohort and among those with PTEN mutations is illustrated in Figure 2. Subjects with a PTEN mutation had an average OFC Z score that was higher than those without a mutation (4.3 ± 1.3 vs. 2.3 ± 1.6; P < 0.0001). The percentage of subjects with a mutation increased with increasing head size. Mutation rates for OFC Z scores from 2.0 to 3.9 was 5 of 43 (11.6%), whereas rates were 6 of 26 (23.1%) for subjects with OFC Z scores ≥ 4.0. This finding concurs with results from previous studies demonstrating a higher yield of PTEN sequencing in those with marked (Z score ≥ 4.0) macrocephaly.10 However, the largest Z score of 9.4 was observed in a subject with DD without a PTEN mutation. Although none of the subjects with a PTEN mutation had an OFC Z score under 2.0, the identical twin of Subject 2 had a diagnosis of ASD, carried the same E157G change, and had an OFC Z score of 0.99.

Distribution of occipital-frontal circumference (OFC) Z scores among subjects. Top, distribution in PTEN-negative subjects; Middle, distribution in PTEN-positive subjects; Bottom, distribution among all subjects.
Comparisons of problems reported in the medical history or findings on physical exam were made between those subjects with a PTEN mutation and those without a mutation. No differences were noted between the groups for the presence of birth defects or reported history of problems in the visual, central nervous system, respiratory, gastrointestinal, genitourinary, musculoskeletal, dermatologic, or immune systems.
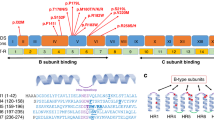
The spectrum of PTEN mutations identified included four missense, three nonsense, three splice-site, and one frameshift mutation. Three of the mutations were paternally inherited, one was maternally inherited, and four were de novo. In three subjects, the inheritance was unknown because parental testing was not performed.
DISCUSSION
We report on a cohort of 114 patients selected for ASD, DD/MR, and/or macrocephaly that had clinical PTEN gene sequencing. We identified mutations in 11 patients (9.6%). We found that 5 of 60 (8.3%) patients with a confirmed ASD were PTEN mutation positive. Further, we found that PTEN mutations were identified in 6 of 49 (12.2%) of patients sequenced for an indication of DD/MR without ASD. Taken together, these data illustrate that PTEN sequencing has a high diagnostic yield when performed in carefully selected populations.
Similar to others, we did not observe a genotype–phenotype correlation in this study. But, like Lachlan et al.5 macrocephaly was a consistent finding. We noted that no PTEN mutations were found in subjects with a normal OFC, with the exception of an identical twin with a diagnosis of autism who had relative, but not absolute, macrocephaly. Although our findings suggest that PTEN testing should be considered in cases of autism and macrocephaly, other syndromes, including fragile X, Sotos syndrome, neurofibromatosis Type 1, and 22q13 deletions may also account for some cases, and multiple causes of brain overgrowth may predispose to autism.12
Various phenotypes, including macrocephaly or macrosomia and DD (without other features of BRRS), VATER-like and Proteus-like phenotypes, have been associated with PTEN mutations.5 The clinical features in our subjects were quite variable and presentations differed significantly even within families. Although we observed unusual skin findings in our patient sample, including café au lait spots, hemangiomata, and a verrucuous lesion, these are not the traditional hamartomatous lesions most commonly described in CS/BRRS. None of the subjects with a PTEN mutation fulfilled diagnostic criteria for a PTEN-related syndrome before testing. In some, such as Patients 7 and 11, this may be because they were evaluated at a very young age before clinical features of CS or BRRS became apparent.
Interestingly, in two of our patients with known pathogenic PTEN mutations (L139X, R130X) the mutation was also identified in phenotypically normal fathers. This emphasizes the need for careful examination of relatives to detect features that may be subtle, or those that are not evident from physical examination alone (such as gastrointestinal hamartomas). Others have observed that a milder phenotype may occur in nonprobands with the same mutation.5
Several of the mutations in our cohort have been reported previously, including three nonsense mutations (R130X, L139X, and R233X), which have been observed in CS, BRRS, and CS/BRRS overlap families and are known to be pathogenic.20 L139X has also been observed in a patient with CS and Sjogren syndrome.21 The novel 520insT frameshift mutation in exon 6 results in premature termination in Patient 4 with autism and macrocephaly22 and is also considered pathogenic.
We identified three splice-site mutations in this clinical sample. The IVS6-1G>A mutation (Patient 9 with DD) results in an alteration of a splice acceptor site as does IVS8-2A>G (Patient 11). Both are considered pathogenic. The latter mutation was also found in the father of Patient 11. He exhibited features of BRRS, including macrocephaly, learning disability, a history of gastrointestinal bleeding, and freckling of the glans penis. IVS8-2A>G has also been previously observed as a somatic mutation in glioblastoma multiforme.23 Patient 5, who possesses an IVS6-3C>G, immediately adjacent to the consensus acceptor site, was investigated further. RT-PCR products of the 4F-9R PTEN primer pairs from Patient 5 contained only a single band of the expected ∼1000 bp size, identical to the control. To ensure there was no altered splicing event, we sequenced products from 15 clones. All were identical to the reference cDNA sequence, with no evidence of altered splice site variants. The significance of this variant is, therefore, unclear.
We identified four missense mutations, one of which has been previously reported as a somatic mutation. The R173H mutation in exon 6 (Patient 7) is one of the most commonly mutated residues in individuals with PTEN-related tumors.24 The missense mutation E157G, observed in Patient 2, results in a change in amino acid charge. The region is also highly conserved in species analyzed, including the frog, chicken, rat, mouse, Rhesus monkey, Chimpanzee, and humans, and it was predicted to be “possibly damaging” using the PolyPhen program. Furthermore, the same mutation was found in the proband's symptomatic identical twin who has a diagnosis of an ASD. The G44D mutation, identified in Patient 6, has not been previously reported. This mutation results in a change in amino acid charge and would be expected to alter the protein's secondary structure. It is also conserved in all species analyzed, suggesting it is pathogenic. Finally, for the T202I alteration found in Patient 8, it is unclear whether or not this change would result in deleterious effects. The Polyphen analysis predicted that this would be a benign variation, yet this region was conserved in all species analyzed. Further, this variant was not observed in 800 control alleles analyzed in our laboratory. More data are required to determine if this variant is pathogenic.
The strength of this study lies in the complete capture of all individuals tested for PTEN mutations at our institution. All testing for mutations is performed in our laboratory, and can be tracked through our laboratory information system. A primary limitation of this study includes ascertainment bias. Our cohort includes patients in whom there was a high index of suspicion for PTEN mutations, thereby limiting the generalizability of our findings. Nonetheless, our study provides clues as to the prevalence of PTEN mutations in highly selected cohorts, such as those referred to genetics, developmental pediatric and neurology clinics for evaluation of ASDs or DD/MR and/or macrocephaly.
Another limitation includes the retrospective accumulation of clinical data. Some clinical details were not recorded or provided in sufficient detail. Furthermore, diagnoses may evolve over time. Four of our DD/MR subjects with PTEN mutations were younger than 2.5 years at the time of study, and may develop an ASD in the future. Therefore, a prospective study to further delineate genotype–phenotype correlations will be needed.
In summary, we have identified a high prevalence of PTEN mutations in a selected population of patients with ASDs, DD/MR, and/or macrocephaly. Several of the mutations that we report are newly identified, adding to the body of literature on this topic. As evidence mounts to support the occurrence of PTEN mutations in the population of children with ASDs and macrocephaly, large prospective studies will be needed to elucidate the true prevalence of these mutations. Our findings suggest that PTEN sequencing is warranted in patients with macrocephaly (>2.0 SD), ASDs, and/or DD. Identification of PTEN mutations has implications for genetic counseling of relatives, and future surveillance of patients and family members.
Patient 5 (mutation IVS6–3C>G) has been subsequently reevaluated in clinic at age 8 years 6 months. He now demonstrates freckling of the glans penis suggesting a diagnosis of BRRS, providing additional evidence the PTEN mutation he carries is pathogenic.
REFERENCES
Maehama T, Dixon JE . PTEN: a tumour suppressor that functions as a phospholipid phosphatase. Trends Cell Bio 1999; 9: 125–128.
; Maehama;T, Taylor;GS, Dixon;JE . PTEN and myotubularin: novel phosphoinositide phosphatases. Annu Rev Biochem; 2001; 70: 247–279.
; Barker;KT, Houlston;RS . Overgrowth syndromes: is dysfunctional PI3-kinase signalling a unifying mechanism?. Eur J Hum Genet; 2003; 11: 665–670.
; Zhou;XP, Waite;KA, Pilarski;R, et al. Germline PTEN promoter mutations and deletions in Cowden/Bannayan-Riley-Ruvalcaba syndrome result in aberrant PTEN protein and dysregulation of the phosphoinositol-3-kinase/Akt pathway. Am J Hum Genet; 2003; 73: 404–411.
; Lachlan;KL, Lucassen;AM, Bunyan;D, Temple;IK . Cowden syndrome and Bannayan Riley Ruvalcaba syndrome represent one condition with variable expression and age-related penetrance: results of a clinical study of PTEN mutation carriers. J Med Genet; 2007; 44: 579–585.
; Marsh;DJ, Kum;JB, Lunetta;KL, et al. PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet; 1999; 8: 1461–1472.
; Goffin;A, Hoefsloot;LH, Bosgoed;E, Swillen;A, Fryns;JP . PTEN mutation in a family with Cowden syndrome and autism. Am J Med Genet; 2001; 105: 521–524.
; Parisi;MA, Dinulos;MB, Leppig;KA, Sybert;VP, Eng;C, Hudgins;L . The spectrum and evolution of phenotypic findings in PTEN mutation positive cases of Bannayan-Riley-Ruvalcaba syndrome. J Med Genet; 2001; 38: 52–58.
; Zori;RT, Marsh;DJ, Graham;GE, Marliss;EB, Eng;C . Germline PTEN mutation in a family with Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome. Am J Med Genet; 1998; 80: 399–402.
; Butler;MG, Dasouki;MJ, Zhou;XP, et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet; 2005; 42: 318–321.
; Herman;GE, Butter;E, Enrile;B, Pastore;M, Prior;TW, Sommer;A . Increasing knowledge of PTEN germline mutations: two additional patients with autism and macrocephaly. Am J Med Genet A; 2007; 143: 589–593.
; Buxbaum;JD, Cai;G, Chaste;P, et al. Mutation screening of the PTEN gene in patients with autism spectrum disorders and macrocephaly. Am J Med Genet B Neuropsychiatr Genet; 2007; 144: 484–491.
; Herman;GE, Henninger;N, Ratliff-Schaub;K, Pastore;M, Fitzgerald;S, McBride;KL . Genetic testing in autism: how much is enough?. Genet Med; 2007; 9: 268–274.
; Zankl;A, Molinari;L . ABase—a tool for the rapid assessment of anthropometric measurements on handheld computers. Am J Med Genet A; 2003; 121: 146–150.
; Farkas;LG . Anthropometry of the head and face, 2nd ed. New York: Raven Press 1994.
; Ogden;CL, Kuczmarski;RJ, Flegal;KM, et al. Centers for Disease Control and Prevention 2000 growth charts for the United States: improvements to the 1977 National Center for Health Statistics version. Pediatrics; 2002; 109: 45–60.
; Sunyaev;S, Ramensky;V, Koch;I, Lathe;W III, Kondrashov;AS, Bork;P . Prediction of deleterious human alleles. Hum Mol Genet; 2001; 10: 591–597.
; Yeargin-Allsopp;M, Rice;C, Karapurkar;T, Doernberg;N, Boyle;C, Murphy;C . Prevalence of autism in a US metropolitan area. JAMA; 2003; 289: 49–55.
; Lord;C, Risi;S, Lambrecht;L, et al. The autism diagnostic observation schedule-generic: a standard measure of social and communication deficits associated with the spectrum of autism. J Autism Dev Disord; 2000; 30: 205–223.
; Eng;C . PTEN: one gene, many syndromes. Hum Mutat; 2003; 22: 183–198.
; Raizis;AM, Ferguson;MM, Robinson;BA, Atkinson;CH, George;PM . Identification of a novel PTEN mutation (L139X) in a patient with Cowden disease and Sjogren's syndrome. Mol Pathol; 1998; 51: 339–341.
; Delatycki;MB, Danks;A, Churchyard;A, Zhou;XP, Eng;C . De novo germline PTEN mutation in a man with Lhermitte-Duclos disease which arose on the paternal chromosome and was transmitted to his child with polydactyly and Wormian bones. J Med Genet; 2003; 40: e92.
; Martinez;R, Schackert;HK, von Kannen;S, Lichter;P, Joos;S, Schackert;G . Independent molecular development of metachronous glioblastomas with extended intervening recurrence-free interval. Brain Pathol; 2003; 13: 598–607.
; Tan;WH, Baris;HN, Burrows;PE, et al. The spectrum of vascular anomalies in patients with PTEN mutations: implications for diagnosis and management. J Med Genet; 2007; 44: 594–602.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: The authors declare no conflict of interest.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.geneticsinmedicine.org).
Rights and permissions
About this article
Cite this article
Varga, E., Pastore, M., Prior, T. et al. The prevalence of PTEN mutations in a clinical pediatric cohort with autism spectrum disorders, developmental delay, and macrocephaly. Genet Med 11, 111–117 (2009). https://doi.org/10.1097/GIM.0b013e31818fd762
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e31818fd762
Keywords
This article is cited by
-
PTEN hamartoma tumor syndrome in childhood and adolescence—a comprehensive review and presentation of the German pediatric guideline
Molecular and Cellular Pediatrics (2022)
-
Genetic Testing in Patients with Neurodevelopmental Disorders: Experience of 511 Patients at Cincinnati Children's Hospital Medical Center
Journal of Autism and Developmental Disorders (2022)
-
Considerations on diagnosis and surveillance measures of PTEN hamartoma tumor syndrome: clinical and genetic study in a series of Spanish patients
Orphanet Journal of Rare Diseases (2022)
-
Conditional Pten knockout in parvalbumin- or somatostatin-positive neurons sufficiently leads to autism-related behavioral phenotypes
Molecular Brain (2021)
-
Cross-level analysis of molecular and neurobehavioral function in a prospective series of patients with germline heterozygous PTEN mutations with and without autism
Molecular Autism (2021)
