
Abstract
Purpose: Hereditary long QT syndrome is named for a prolonged QT interval reflecting predisposition to ventricular arrhythmias and sudden death. A high rate in a remote, northern Canadian First Nations community was brought to attention.
Methods: Two severely affected index cases and 122 relatives were ascertained using community-based participatory research principles. Genetic sequencing of five known genes responsible for long QT syndrome was carried out on the index cases, leading to the identification of a novel missense mutation. Functional properties of the identified mutation were studied in transfected mouse ltk- cells using whole cell patch clamp techniques. Corrected QT interval measurements were obtained from participants and subsequent genotyping of relatives was carried out.
Results: In the two index cases, a novel missense mutation (V205M) was identified in the S3 transmembrane helix of KvLQT1, the pore forming domain of the IKs channel complex. In transfected mouse ltk-cells the V205M mutation suppressed IKs by causing a dramatic depolarizing shift in activation voltage coupled with acceleration of channel deactivation. Twenty-two mutation carriers had a significantly higher mean corrected QT interval than noncarriers (465 ± 28 milliseconds vs. 434 ± 26 milliseconds, P < 0.0001); however, 30% of carriers had a corrected QT interval below 440 milliseconds.
Conclusion: A novel KCNQ1 mutation in this founder population likely confers increased susceptibility to arrhythmias because of decreased IKs current. Even with a common mutation within a relatively homogenous population, clinical expression remains variable, exemplifying the multifactorial nature of long QT syndrome, and supporting the difficulty of definitive diagnosis without genetic testing. A community participatory approach enabled a comprehensive evaluation of the impact.
Similar content being viewed by others


Main
Long QT syndrome (LQTS), which can be acquired or congenital, results from delayed repolarization of cardiac ventricular action potentials, and is characterized by a prolongation in the QT interval of the electrocardiogram (ECG). It can be associated with a serious multifocal ventricular tachyarrhythmia, torsades de pointes,1 which may lead to syncope and sudden death. Genetically heterogeneous, at least eight different genes are known to be responsible for the congenital form.2,3 Although considered to be inherited in an autosomal dominant pattern because a single mutation may alter channel function, compound mutations4 and common polymorphisms5 also play a role in the pathogenesis of congenital LQTS, illustrating the complex etiology.6 Genetic predisposition may play an important role in acquired drug-induced LQTS7 and furthermore a number of common medications increase the risk of a cardiac event for those with congenital LQTS.8 Treatment is effective in reducing mortality and consists of avoidance of QT prolonging drugs, the use of beta blockers and/or implanted cardioverter-defibrillators.8,9
Congenital LQTS is relatively rare, affecting an estimated 1 in 5,000–10,000 people,10 but it is disproportionately prevalent in a Canadian First Nations community of 5500 people in northern British Columbia (the Gitxsan), whose ancestors are said to have resided there for thousands of years without European contact until 1860. Forty individuals had been diagnosed as likely having the condition, and another 200 were considered at risk. Although founder mutations have been previously reported in Finland11 and South Africa,12 hereditary LQTS has not been reported in North American Aboriginal populations. Although a report from the Strong Heart Study found that prolonged corrected QT interval (QTc) was an independent predictor for all causes of mortality for a large American Indian cohort, congenital LQTS per se was not discussed.13
Through a community initiated study of 124 participants identified because they either had LQTS or had a relative diagnosed with LQTS, we identified a novel missense mutation (valine substitution with methionine at position 205, V205M) in the S3 transmembrane region of KCNQ1, the α-subunit for the slow delayed rectifier potassium channel, IKs. The V205M mutation was present in 22 individuals in three separate kindreds who are of the same First Nation, suggesting a frequency of at least 1 in 250 people in this remote community.
Four KvLQT1 subunits coassemble with MinK accessory subunits to form the slowly activating and slowly deactivating IKs in the heart, which modulates the repolarization of cardiac action potentials,14,15 particularly during sympathetic activation and tachycardia.16 LQTS linked to mutations in the KCNQ1 gene is usually attributed to a decrease or suppression of IKs by the prevention of efficient protein folding and trafficking to the cell membrane,17–20 and less often to modification of the response of IKs channels to changes in voltage such that their contribution to repolarization is diminished.21–23
Aboriginal communities may be hesitant to participate in genetics research because of past research practices which may not have considered the needs of the community.24 This study demonstrates the merits of carrying out genetics research in a community-based participatory research manner,25 one that was mutually acceptable to the community and to the researchers.
MATERIALS AND METHODS
Study participants
Consistent with the principles of community-based participatory research,26 community members alerted university researchers to the need to investigate a high rate of LQTS. A local research advisory committee consisting of lay community members and medical personnel was struck under the authority of the Gitxsan Health Society to advise and govern the research. Research protocols were developed in a participatory manner. Community approval and individual consents were obtained, following University of British Columbia and Northern Health Authority ethics approval. After clinical assessment of two index cases, family members were invited to participate. If not previously carried out, an ECG was done and all ECGs on file were collected for QTc measurements. Genetic Counselors (R.R./S.M.) recorded family histories and obtained medical records. Blood or saliva was collected for DNA extraction under the stipulation of DNA on Loan.24
QTc measurements
For QTc measurements, 12-lead ECG information was used. Analysis was carried out by a cardiologist (S.S.) blinded to the clinical and mutation status of the participant. QTc intervals were determined in all leads using Bazett correction,27 to ensure that representative measurements were being made, but only lead II was used for the analysis. For sinus arrhythmia, the QT interval was taken using an interval which followed an RR interval approximating the average heart rate. The end of the T wave was defined using published criteria.28 For individuals with more than one ECG recording, an average QTc was calculated from their individual ECG records, and this value was used in the determination of the mean QTc of the mutation “carrier” and “noncarrier” groups. However, all QTc determinations are shown graphically. Statistical analysis for comparisons between groups included 95% confidence intervals and Student t test, with a value of P < 0.05 considered significant. Odds ratios were used when appropriate.
Sequencing of genes and genotyping
DNA was extracted from blood or saliva using standard methods from 124 participants (all samples available). For the two index cases, the entire coding regions of SCN5A, KCNQ1, KCNH2, KCNE1, and KCNE2 (62 exons) were polymerase chain reaction (PCR) amplified based on primers and conditions previously published29,30 and sequenced on an ABI 3730xl Analyzer (J.E.). Dimethyl sulfoxide or betaine was added when needed to improve PCR amplification.31 Genotyping of the identified mutation was performed using a custom Taqman SNP genotyping assay from Applied Biosystems (Foster City, CA) on an ABI Prism 7000 Sequence detection system in the clinically accredited laboratory of one of the investigators (B.C.).
Molecular biology
KCNQ1 and KCNE1 genes were purchased from Origene Technologies (Rockville, MD). The missense mutation V205M within the third transmembrane (S3) region of the KCNQ1 gene was constructed using a two-step PCR reaction.
Cell preparation and transfection
Electrophysiology was carried out on transiently transfected mouse ltk- cells. One microgram of KCNQ1 and 2.5 μg of KCNE1 accessory subunit DNA was incubated with 1 μg of pGFP and 3 μL of LipofectAMINE 2000 (Gibco-BRL) in serum-free OPTI-MEM (Gibco-BRL), and then added to the dishes containing cells in modified Eagle's medium with 10% fetal bovine serum. Cells were allowed to grow overnight before recording.
Solutions and electrophysiological procedures
Standard patch clamp procedures on cultured cells were used. Patch pipettes contained (in mM): KCl, 130; EGTA, 5; MgCl2, 1; HEPES, 10; Na2ATP, 4; GTP, 0.1; and was adjusted to pH 7.2 with KOH. The bath solution contained (in mM): NaCl, 135; KCl, 5; HEPES, 10; sodium acetate, 2.8; MgCl2, 1; CaCl2, 1; and was adjusted to pH 7.4 with NaOH. Throughout the text, data are presented as mean ± SEM. Significance was tested using Student unpaired t test or one-way analysis of variance as appropriate and a value of P < 0.05 was considered significant.
RESULTS
Study population demographics
Index case No. 1 was 38 years at presentation, and had been resuscitated from an apparent cardiac arrest which occurred during a competitive sports activity. She had a previous history of syncope and palpitations. Index case No. 2 also had a history of frequent palpitations, with brief episodes of altered consciousness and weakness. A prolonged QTc was noted at age 37. Both were managed with implanted defibrillators. Although both were Gitxsan from the same community, there was no knowledge of being related within four generations known to them (Fig. 1 A andB). They were both found to have the V205M mutation as a result of a G to A substitution in exon 4 of the KCNQ1 gene (Fig. 1C), a mutation predicted to be pathogenic. Both of the index cases also had a polymorphism in KCNE1, G38S, which is not thought to modify the QT interval.32 No other mutations were detected in both index cases in the three other LQTS genes screened (SCN5A, KCNH2, KCNE2), although a number of polymorphisms in each were detected. One hundred twenty-two additional community participants were genotyped for the presence of V205M KCNQ1. Twenty relatives carried the V205M mutation (total 22) and 102 were without the mutation. One additional kindred, not known to be related to the other two, but with affected individuals harboring the V205M mutation, is not shown. Of the 22 individuals with mutations, eight were previously considered definitively affected by their physicians, four were considered “borderline,” and six were considered unlikely to have a mutation. Four had not been previously assessed. Of the 102 individuals without mutations, one individual was thought to be affected by her clinician after presenting with a moderately elevated QTc and palpitations. For that individual, in addition to genotyping for the V205M mutation, the coding regions of SCN5A, KCNQ1, KCNH2, KCNE1 and KCNE2 (62 exons) were sequenced as was carried out for the two index cases. Although no mutations were detected, one common polymorphism (SCN5A H558R), thought to occasionally predispose to Brugada-like ECG changes,33 was found.

Simplified family pedigree of index case families, representing two of three kindreds from the same community with the V205M mutation. A, Index case No. 1 extended family. B, Index case No. 2 immediate family. The index cases are identified with arrows. Presence of V205M mutation confirmed by genotyping is designated by the filled symbols, absence of the V205M mutation is designated by a horizontal line through the symbol. Unexplained death is designated with half-filled symbols with oblique line designating death. C, DNA sequencing of KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2 genes of the index cases revealed a G to A substitution in exon 4 of the KCNQ1 gene that results in mutation of valine 205 to methionine.
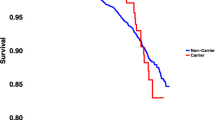
Of the 22 with V205M mutations, 21 had available ECGs for review (total 78) and of the noncarriers, 64 had available ECGs (total 86) (Fig. 2A). The age range for those with the mutation was 18–69 years (mean 44.3), and for those without the mutation was 3–81 years (mean 33.4). There were 20 individuals without mutations who were younger than 18 years. There was no difference in the QTc between those below 18 years and those above (431 ± 3 milliseconds vs. 437 ± 4 milliseconds, P = 0.33). Of those with the mutation, 77% were women compared with 56% without (OR, 2.2; 95% CI, 0.9–5.6). There was no difference between the QTc of male and female noncarriers (430 ± 4 milliseconds vs. 437 ± 4 milliseconds, P = 0.13, respectively). There was no difference between the male carriers and noncarriers (430.3 ± 10 milliseconds vs. 430.1 ± 4 milliseconds, P = 0.98) but there was a significant difference between male and female carriers (430.3 ± 10 milliseconds vs. 470.9 ± 6 milliseconds, P = 0.02). Adult females with mutations (n = 18) were also compared with adult females without mutations (n = 40) and found to have the most significant difference in QTc measurement (471 ± 6 milliseconds vs. 438 ± 4 milliseconds, P < 0.0001). Although all mutation carriers had a significantly higher mean QTc than all noncarriers (465 ± 28 milliseconds vs. 434 ± 26 milliseconds, P < 0.0001) (Fig. 2A), 30% of carriers had at least one QTc below 440 milliseconds. Figure 2B depicts eight cases with more than five ECGs, demonstrating substantial intrapatient variability for QTc. Although a QTc of >430 milliseconds had a sensitivity for mutation positive status of 88%, the specificity was only 46%. On the other hand, a QTc of >466 milliseconds conferred a specificity of 86%, but had <50% sensitivity.

The V205M mutation prolongs the QTc. A, QTc intervals determined at rest for mutation carriers and noncarriers. Each point represents one QTc determination. Measurement of QTc was carried out on 78 ECGs from 21 carriers and 86 ECGs from 64 noncarriers, as described in “Methods.” The short horizontal bars represent the mean QTc of individuals which were 465 ± 4 milliseconds and 434 ± 3 milliseconds, in carriers and noncarriers, respectively. QTc variations among eight V205M carriers are shown in (B).
The V205M mutation alters activation and deactivation properties of IKs channels
Wt and mutant KCNQ1 channels were coexpressed with the accessory β-subunit KCNE1 in ltk- cells, as they possess no endogenous K+ channel activity. The responses of Wt and mutant IKs channels to voltage were characterized and compared by recording ionic currents at 35°C from cells expressing Wt KCNQ1+KCNE1 (Fig. 3A) and V205M KCNQ1+KCNE1 (Fig. 3B) during 2 second depolarizations. Outward currents were recorded positive to −10 mV in the Wt channels (Fig. 3C), but strikingly, current activation was slowed and shifted to more depolarized potentials in the presence of the V205M mutation, appearing positive to +20 mV (Fig. 3C). The voltage dependence of activation was determined by measuring the half-activation voltages (V1/2) for Wt and V205M channels, and was shifted from +10.4 to +44.1 mV in the V205M channels.

The V205M mutation results in a depolarizing shift in the voltage dependence of IKs activation. Wt KCNQ1+KCNE1 (A) and V205M KCNQ1+KCNE1 (B) ionic currents were recorded at 35°C during 2 seconds depolarizing pulses to between −50 mV and +70 mV followed by a 2 seconds repolarizing pulse to −50 mV. C, g-V relationships were constructed by plotting the peak tail currents at −50 mV and the data were fitted to a single Boltzmann function. The V1/2's of activation were 10.4 ± 3.2 mV (n = 7) and 44.1 ± 5.0 mV (n = 6) with slope factors of 8.5 ± 0.7 mV and 9.9 ± 1.1 mV for Wt KCNQ1+KCNE1 (n = 7) and V205M KCNQ1+KCNE1 (n = 6), respectively. D, Ionic currents were recorded from cells expressing Wt KCNQ1+KCNE1 and V205M KCNQ1+KCNE1 during 2 seconds depolarizing pulses to + 80 mV at 35°C followed by repolarizing pulses to potentials between −40 mV and −100 mV in 10 mV increments.
In addition to modulating IKs channel activation properties, the V205M mutation accelerated the rate of channel deactivation (Fig. 3D, tail currents). It is clear that the V205M mutation accelerated deactivation at all the potentials tested compared with Wt channels. These data suggest that the V205M mutation reduces IKs channel availability by shifting the voltage dependence of activation to more depolarized potentials, in addition to slowing the rate of activation and accelerating the rate of channel deactivation.
DISCUSSION
The majority of KCNQ1 mutations typically produce a loss of function by disrupting protein folding, assembly, and/or trafficking to the membrane.17–20 In contrast, the V205M mutation exhibited markedly slowed activation and accelerated deactivation (Fig. 3). The half-activation potential was depolarized >30 mV without altering the slope factor. This impairs the physiological ability of IKs channels to respond to rapid heart rhythms by beat-to-beat summation of current levels and may lead to a reduction of the overall repolarization reserve in affected hearts.34 Similar electrophysiological findings (a depolarizing shift of activation) have been reported for the Q357R mutation in the S6 region,21 R294H in S4,22 and R555C in the C-terminus,23 but the effect of the V205M mutation on the kinetics of IKs channels reported here is much more severe. Although four other mutations (D202H, I204M, S209F and V215M) have been detected in the S3 region of KCNQ1 genes of patients with LQTS,35 none of these mutations have yet been characterized. Any reduction of IKs because of inherited mutations in the KCNQ1 genes could result in an increased risk for ventricular arrhythmia during periods of elevated sympathetic activity such as exercise and emotion1 consistent with the findings in index case No. 1, who experienced cardiac arrest during an episode of emotional stress while exercising.
This is the first description of a founder mutation causing LQTS in a North American Aboriginal community, and as in other groups, the diagnosis of LQTS in family members of affected individuals presented a significant clinical challenge.36,37 Here, it was exacerbated because of a large number of individuals who had QTc measurements in the borderline range with 30% of carriers having at least one QTc below 440 milliseconds. Although it has been suggested that a QTc of 430 milliseconds is a reasonable screening cut-off for diagnosing affected relatives of mutation carriers with acceptable sensitivity and specificity,37 this was not true in our relatively homogenous population. The specificity at that QTc was <50% for our population, suggesting a baseline increased QTc in nonmutation carriers. Although not specifically studied in this community, a baseline increase in QTc may be associated with obesity and diabetes, both common in North American Native populations.13 In this study, the highly significant difference in QTc between mutation positive and negative participants, and especially mutation positive and negative women, clinically supports the in vitro confirmation of pathogenesis, but may not be helpful for individual clinical assessments of those at risk.
The paucity of men with mutations in our study is consistent with other observations,38 and may be a result of transmission distortion39 or ascertainment bias in that women may be more severely clinically affected,38 and more apt to enter a study such as this. It is also possible that men at high risk are more hesitant to learn their mutation status. Several men in the community known to be at 50% risk have not come forward for testing. The fact that there are so few mutation positive men in our study limits any assumptions that men with mutations are less likely to have an elevated QTc, although our preliminary results suggest that possibility. More research on the impact of KCNQ1 V205M on men in this community is indicated.
Although QTc length may be helpful in predicting risk of a cardiac event in LQTS,40 the marked intrapatient variability with resting ECG in our mutation carriers illustrates the difficulty in interpreting mutation status and subsequent risk based on any single QTc measurement. In reviewing the number of unexpected deaths apparent in these families, and in reviewing the case studies of those who have presented symptomatically, the seriousness of the presence of the V205M KCNQ1 mutation as a predisposition to LQTS cannot be ignored, although the presence of this mutation alone may be insufficient to declare that an individual has LQTS. More research is needed to determine whether functional polymorphisms in KCNQ1 and other genes responsible for LQTS might contribute to the variable presentation evident in this community, and typically seen in families of those with LQTS.4–6 Nongenetic mechanisms underlying the variability in expression of the condition, also need to be considered.41
Aboriginal communities may hesitate to participate in genetic research that will not have a direct impact on the health of their community.24 This community recognized that addressing LQTS was a health care priority, directed the initiation of this study, and has continued to be involved in the implementation and governance. A community-based approach enabled the research42 and is in keeping with the current Canadian Institutes for Health Research Guidelines for Health Research Involving Aboriginal People.43
The impact of LQTS in this community was profound in that local health care providers and consultant cardiologists were often unable to predict which community members were at risk for arrhythmias, resulting in general population avoidance of QT prolonging medications, many of which are commonly used in day to day clinical practice. Furthermore, several individuals were being treated medically on speculation that their borderline QTc was reflective of congenital LQTS, especially if they had close relatives with LQTS. Because the closest tertiary care center (Vancouver) is 1200 km from Hazelton, defining the mutation and confirming pathogenesis has provided a valuable and inexpensive diagnostic test for those with features of LQTS and who are related to mutation carriers. Nonetheless, clinical monitoring of those with the mutation will remain a challenge in this remote community. To date, all mutation carriers have been referred to a cardiologist for assessment of the need for medical or interventional therapy. Continued monitoring is through the local hospital and health centers. Furthermore, portable automatic defibrillators have now been made available throughout the community. Further community education strategies are planned.
Although 22 individuals with V205M mutations have been identified to date, suggesting a rate of at least 1/250 definitively affected individuals in this community, the expanded family histories with added information of mutation status reveal that at least 40 more individuals are at 50% risk of having a mutation and another 80 are at 25% risk. This suggests the mutation positive rate could reach higher than 1/100 in this and surrounding communities. The broader community impact will become known once random community screening for the mutation is implemented. Newborn screening has also been considered,44 but population specific evidence of increased risk of death in infancy and early childhood will inform that decision. Ongoing monitoring of those with and without mutations will shed light on the comparative risk of sudden death, and of the merits of treating mutation carriers with beta blockers or defibrillators.
A community-based approach to the study of LQTS in a northern British Columbia First Nations community has led to the identification of a novel LQTS mutation, V205M KCNQ1, and an understanding of its pathogenesis. The delineation of this founder mutation will now allow definitive diagnosis of a predisposition to LQTS and appropriate management to reduce the risk of serious cardiac events. Ongoing studies will provide further insight into variable clinical expression, while optimum screening and management strategies are explored to further benefit this community.
References
Keating MT, Sanguinetti MC . Molecular and cellular mechanisms of cardiac arrhythmias. Cell 2001; 104: 569–580.
Ackerman MJ . Cardiac channelopathies: it's in the genes. Nat Med 2004; 10: 463–464.
Modell SM, Lehmann MH . The long QT syndrome family of cardiac ion channelopathies: a HuGE review. Genet Med 2006; 8: 143–155.
Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC . Compound mutations: a common cause of severe long QT syndrome. Circulation 2004; 109: 1834–1841.
Schulze-Bahr E . Arrhythmia predisposition: between rare disease paradigms and common ion channel gene variants. J Am Coll Cardiol 2006; 48: A67–A78.
Gargus JJ . Unravelling monogenic channelopathies and their implications for complex polygenic disease. Am J Hum Genet 2003; 72: 785–803.
Abbott GW, Sesti F, Splawski I, et al. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 1999; 97: 175–187.
Schwartz P . The congenital long QT syndromes from genotype to phenotype: clinical implications. J Intern Med 2006; 259: 39–47.
American College of Cardiology, American Heart Association Task Force, European Society of Cardiology. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death—executive summary: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines. Circulation 2006; 114: 1088–1132.
Chiang C . Congenital and acquired long QT syndrome: current concepts and management. Cardiol Res 2004; 12: 222–234.
Piippo K, Swan H, Pasternack M, et al. A founder mutation of the potassium channel KCNQ1 in long QT syndrome: implications for estimation of disease prevalence and molecular diagnostics. J Am Coll Cardiol 2001; 37: 562–568.
Brink PA, Crotti L, Corfield V, et al. Phenotypic variability and unusual clinical severity of congenital long-QT syndrome in a founder population. Circulation 2005; 112: 2602–2610.
Okin PM, Devereux RB, Howard BV, Fabsitz RR, Lee ET, Welty TK . Assessment of QT interval and QT dispersion for prediction of all-cause and cardiovascular mortality in American Indians: the Strong Heart Study. Circulation 2000; 101: 61–66.
Sanguinetti MC, Curran ME, Zou A, et al. Coassembly of KvLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature 1996; 384: 80–83.
Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G . KvLQT1 and IsK (minK) proteins associate to form the IKs cardiac potassium current. Nature 1996; 384: 78–80.
Romey G, Attali B, Chouabe C, et al. Molecular mechanism and functional significance of the MinK control of the KvLQT1 channel activity. J Biol Chem 1997; 272: 16713–16716.
Shalaby FY, Levesque PC, Yang WP, et al. Dominant-negative KvLQT1 mutations underlie the LQT1 form of long QT syndrome. Circulation 1997; 96: 1733–1736.
Dahimene S, Alcolea S, Naud P, et al. The N-terminal juxtamembranous domain of KCNQ1 is critical for channel surface expression: implications in the Romano-Ward LQT1 syndrome. Circ Res 2006; 99: 1076–1083.
Huang L, Bitner-Glindzicz M, Tranebjaerg L, Tinker A . A spectrum of functional effects for disease causing mutations in the Jervell and Lange-Nielsen syndrome. Cardiovasc Res 2001; 51: 670–680.
Bianchi L, Priori SG, Napolitano C, et al. Mechanisms of IKs suppression in LQT1 mutants. Am J Physiol Heart Circ Physiol 2000; 279: H3003–H3011.
Boulet IR, Raes AL, Ottschytsch N, Snyders DJ . Functional effects of a KCNQ1 mutation associated with the long QT syndrome. Cardiovasc Res 2006; 70: 466–474.
Chouabe C, Neyroud N, Richard P, et al. Novel mutations in KvLQT1 that affect Iks activation through interactions with Isk. Cardiovasc Res 2000; 45: 971–980.
Chouabe C, Neyroud N, Guicheney P, Lazdunski M, Romey G, Barhanin J . Properties of KvLQT1 K+ channel mutations in Romano-Ward and Jervell and Lange-Nielsen inherited cardiac arrhythmias. EMBO J 1997; 16: 5472–5479.
Arbour L, Cook D . DNA on loan: issues to consider when carrying out genetic research with Aboriginal families and communities. Community Genet 2006; 9: 153–160.
Macaulay AC, Commanda LE, Freeman WL, et al. Participatory research maximizes community and lay involvement. BMJ 1999; 319: 774–778.
Israel B, Schulz AJ, Parker EA, Becker AB . Community based participatory research: policy recommendations for promoting a partnership approach in health research. Educ Health 2001; 14: 182–197.
Bazett HC . An analysis of the time-relations of electrocardiograms. Heart 1920; 7: 353–370.
Lepeschkin E, Surawicz B . The measurement of the Q-T interval of the electrocardiogram. Circulation 1952; 6: 378–388.
Splawski I, Shen J, Timothy KW, Vincent GM, Lehmann MH, Keating MT . Genomic structure of three long QT syndrome genes: KVLQT1, HERG, and KCNE1. Genomics 1998; 51: 86–97.
Syrris P, Murray A, Carter ND, McKenna WM, Jeffery S . Mutation detection in long QT syndrome: a comprehensive set of primers and PCR conditions. J Med Genet 2001; 38: 705–710.
Musso M, Bocciardi R, Parodi S, Ravazzolo R, Ceccherini I . Betaine, dimethyl sulfoxide, and 7-deaza-dGTP, a powerful mixture for amplification of GC-rich DNA sequences. J Mol Diagn 2006; 8: 544–550.
Akyol M, Jalilzadeh S, Sinner MF, et al. The common non-synonymous variant G38S of the KCNE1-(minK)-gene is not associated to QT interval in Central European Caucasians: results from the KORA study. Eur Heart J 2007; 28: 305–309.
Makielski J, Ye B, Valdivia CR, et al. A ubiquitous splice variant and a common polymorphism affect heterologous expression of recombinant human SCN5A Heart Sodium Channel. Circ Res 2003; 93: 821–828.
Jost N, Virág L, Bitay M, et al. Restricting excessive cardiac action potential and QT prolongation: a vital role for IKs in human ventricular muscle. Circulation 2005; 112: 1392–1399.
Napolitano C, Priori SG, Schwartz PJ, et al. Genetic testing in the long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice. JAMA 2005; 294: 2975–2980.
Priori SG, Napolitano C, Schwartz PJ . Low penetrance in the long-QT syndrome: clinical impact. Circulation 1999; 99: 529–533.
Hofman N, Wilde AA, Kaab S, et al. Diagnostic criteria for congenital long QT syndrome in the era of molecular genetics: do we need a scoring system?. Eur Heart J 2007; 28: 575–580.
Kaab S, Pfeufer A, Hinterseer M, Nabauer M, Schulze-Bahr E . Long QT syndrome: why does sex matter?. Z Kardiol 2004; 93: 641–645.
Imboden M, Swan H, Denjoy I, et al. Female predominance and transmission distortion in the long-QT syndrome. N Engl J Med 2006; 355: 2744–2751.
Roden DM . Long-QT syndrome. N Engl J Med 2008; 358: 169–176.
Schwartz PJ, Vanoli E, Crotti L, et al. Neural control of heart rate is an arrhythmia risk modifier in LQTS. J Am Coll Cardiol 2008; 51: 920–929.
Flicker S, Savan B, Kolenda B, Mildenberger M . A snapshot of community-based research in Canada: who? what? why? how?. Health Educ Res 2008; 23: 106–114.
Canadian Institutes of Health Research. CIHR guidelines for health research involving aboriginal people. Ottawa, Ontario; 2007. Available at: http://www.cihr-irsc.gc.ca/e/29134.html. Accessed April 8, 2008.
Tester DJ, Ackerman MJ . Sudden infant death syndrome: how significant are the channelopathies?. Cardiovasc Res 2005; 67: 388–396.
Acknowledgements
This work was supported by the Canadian Institutes of Health Research (CIHR) and the Heart and Stroke Foundation of British Columbia and Yukon, and the research scientists are funded through the Heart and Stroke Foundation of British Columbia and Yukon (to D.F. and S.R.) and the Michael Smith Foundation for Health Research (to L.A.).
We thank the Gitxsan research advisory committee including Myrtle Goertzen, Julia Sundell, Neil Belanger, and the larger community for their involvement; the health care providers of the Wrinch Memorial Hospital, Sarah McIntosh, Lee-Anna Huisman, Drs. Stanley Tung and Charles Kerr for their clinical contributions to the study; Ms. Ka Kee Chiu for preparation of cells and Ms. Kyung Hee Park for cell and sequencing preparation.
The Gitxsan Health Society Board of Directors and the research advisory committee reviewed the manuscript before publication.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Arbour, L., Rezazadeh, S., Eldstrom, J. et al. A KCNQ1 V205M missense mutation causes a high rate of long QT syndrome in a First Nations community of northern British Columbia: a community-based approach to understanding the impact. Genet Med 10, 545–550 (2008). https://doi.org/10.1097/GIM.0b013e31817c6b19
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e31817c6b19
Keywords
This article is cited by
-
Genomics and inclusion of Indigenous peoples in high income countries
Human Genetics (2023)
-
Epidemiology of inherited arrhythmias
Nature Reviews Cardiology (2020)
-
Inherited cardiac arrhythmias
Nature Reviews Disease Primers (2020)
-
7DHC-induced changes of Kv1.3 operation contributes to modified T cell function in Smith-Lemli-Opitz syndrome
Pflügers Archiv - European Journal of Physiology (2016)
-
Drug-induced QT interval prolongation in cancer patients
Oncology Reviews (2010)
