
Abstract
Although the pace of gene discovery for rare genetic diseases has accelerated during the past decade, in part, due to the success of the Human Genome Project, translation of these discoveries to clinical utility has lagged behind. In particular, identification of the gene responsible for a Mendelian disorder immediately presents the opportunity for molecular genetics diagnostics to confirm clinical diagnoses, provide accurate genetic counseling information and recurrence risks, as well as carrier testing and prenatal diagnosis opportunities for families. To move these discoveries from a research setting to clinical utility, we describe two successful models for partnerships between research laboratories with Clinical Laboratory Improvement Amendments-certified clinical molecular diagnostic laboratories. Contrary to common misconceptions, molecular genetic testing for very rare diseases can be performed in a high-quality clinical setting in a financially self-sustaining or even profitable manner. Key elements to the success of these models include a Clinical Laboratory Improvement Act-certified diagnostic laboratory with a commitment to very rare genetic disease testing, active involvement of genetic counselors and clinical geneticists, and partnerships with research experts and patient support groups specific to each disease.
Similar content being viewed by others

DEFINITION OF LABORATORIES
Clinical Laboratory Improvement Amendments (CLIA)-certified clinical molecular diagnostic laboratory—a molecular diagnostic laboratory that has obtained formal certification through CLIA,1 under the oversight of Centers for Medicaid and Medicare Services, to perform molecular genetic testing of patient samples with appropriate quality control/quality assurance procedures in place. This will also be referred to as CLIA-certified diagnostic laboratory or diagnostic laboratory in the text.
Research laboratory—a laboratory whose primary focus is on research of genetic disorders and does not have CLIA certification mandated in the United States for any laboratory testing of patient samples in which results may be communicated to physicians or patients.
BACKGROUND AND HISTORY
Ultra rare or orphan genetic disorders are defined as diseases that affect <2000 people in the United States; however, when taken as a group affect approximately one in 12 individuals (25 million) in the United States (http://rarediseases.info.nih.gov/). Several national committees have addressed the issues of access to quality genetic testing for ultra rare or orphan genetic diseases.2–4 Despite these deliberations, there were not any organized or proactive efforts by professional societies, federal agencies, or advocacy groups to improve access to genetic testing for these disorders until very recently (see Ledbetter and Faucett, this issue page 309).
Many individual researchers have felt a strong personal obligation to provide mutation analysis for diagnosis, carrier detection, and even prenatal diagnosis for the families that have participated in their research. Such testing has often been unavailable due to the absence of CLIA-certified diagnostic laboratories where the expertise and resources to provide such testing exists. In addition, research laboratories are not generally set up to perform diagnostic testing as the appropriate quality control measures are not in place leading to more susceptibility to error (e.g., sample mix-up), long turnaround time, and limitations in the types of assays that can be performed. Genetic testing performed in CLIA-certified diagnostic laboratories on the other hand is more accurate, performed in a timely manner and more amenable to different types of genetic testing.
Our own experience led each of us to become involved in providing such genetic testing for disorders initially from our own research laboratories, but we decided early on to move this testing into a CLIA-certified clinical diagnostic laboratory setting. We independently developed models for “niche diagnostics” of ultra rare genetic diseases. It has proven possible to not only fill a critical void in access to genetic testing but has been shown to be cost-effective when a single laboratory serves as a reference laboratory for all testing for even a very rare disorder (e.g., lissencephaly). In addition, this centralized approach creates a much larger data set and experience for a diagnostic “Center of Excellence” in collaboration with research experts and clinical experts for each disorder.
TEST TRANSLATION
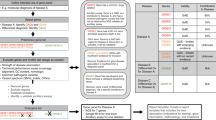
The process of gene discovery and the identification of the molecular basis of genetic disorders have not translated quickly or easily into diagnostic testing for these disorders. There has generally been a lack of interaction between the research and CLIA-certified clinical molecular diagnostic laboratories to move these findings into the diagnostic arena, often leaving patients and their families in a position where genetic testing for their disorder is not accessible (Fig. 1).

Flowchart depicting the common process of gene discovery and the lack of appropriate translation to genetic testing.
One approach to test translation is for a research laboratory to undergo CLIA certification to enable them to perform molecular genetic testing for disorders of research interest. A recent successful example of this is that of comprehensive molecular testing for breast cancer related genes in a research laboratory that had obtained CLIA-certification.5 However, this is a very time-consuming and laborious process for a research laboratory to undergo and may not be an appropriate expenditure of research grant-funding.
An alternative approach to test translation is to form a collaborative effort between a research laboratory and an existing CLIA-certified clinical molecular diagnostic laboratory. This was the approach taken at the University of Chicago in relation to the developmental brain disorder lissencephaly. With the identification of different genes implicated in lissencephaly and the characterization of mutations in these genes in patients, information was available that was of direct benefit to patients and their families. Because of the rarity of the disorder and the lack of any common mutations in patients, genetic testing for this group of disorders was not available. It was decided at that time to form a collaborative effort between the research and CLIA-certified clinical molecular diagnostic laboratories within the institution and establish a system to provide patients with results obtained through research studies in a manner that followed appropriate quality control guidelines. This was followed soon thereafter by collaborative efforts between research laboratories at other institutions and the University of Chicago CLIA-certified diagnostic laboratory to develop testing for other ultra rare genetic disorders. Experience has shown that effective and high quality test translation requires a close relationship and good communication between the research and diagnostic laboratories.
A related approach is to set up a CLIA-certified clinical molecular diagnostic laboratory with a focus on ultra rare genetic disorders. This was the approach taken by GeneDx. Inc. GeneDx, Inc. is a small clinical laboratory that was started by two long-time National Institutes of Health (NIH) scientists, whose careers had focused on understanding the clinical and genetic aspects of rare dermatological disorders, including congenital recessive ichthyoses and epidermolytic ichthyoses. Frustrated by the constraints on research laboratories from providing mutation information to the participating families, by the difficulty of running a CLIA-certified diagnostic laboratory component within the research laboratory, and by the increasing requests from referring physicians and patients themselves for mutation testing in their affected children, carrier testing of relatives, and prenatal diagnosis, for which resources were not available in their laboratory, the investigators left the NIH and started a private laboratory company to provide these services.
VIABILITY OF ULTRA RARE GENETIC DISEASE TESTING
A natural thought by CLIA-certified clinical molecular diagnostic laboratories when asked to set up testing for an ultra rare genetic disease is that it is not cost-effective. Each disease is very rare and only a handful of patients will require the test. Performing the test can be very expensive, especially if the gene is large. As no common mutations are usually present in these disorders, it is necessary to sequence or perform some mutation scanning method for the entire gene. Diagnostic laboratories tend to set up testing for more common disorders or those in which the test can be targeted toward a specific/set of specific mutations. This is particularly true for the larger diagnostic laboratories that are designed to test large numbers of samples in a batch format and have protocols that minimize cost, enabling tests such as that for cystic fibrosis, fragile X, sickle cell disease, etc., to be performed in a very cost-effective manner. It is often very difficult for smaller diagnostic laboratories, especially academic laboratories, to compete against these larger laboratories with these types of tests.
Testing for rare orphan genetic disorders presents an opportunity for academic laboratories and small commercial entities to provide “niche diagnostic” testing for which competition from the larger laboratories is not likely to be present. There is both a need and an obligation to provide testing for ultra rare orphan genetic diseases as the genetic basis of these disorders become understood.
The technology for diagnostic testing for all these disorders is primarily the same, i.e., DNA sequencing, possibly preceded by other mutation scanning methods for the detection of mutations. The experience of the University of Chicago (an example of an academic laboratory) and of GeneDx (an example of a small commercial enterprise), where the focus of the laboratories are on genetic testing for rare orphan genetic diseases, is that such testing can be done in a CLIA-certified molecular diagnostic laboratory setting under rigorous standards and is financially viable and may even be profitable.
MODELS OF TEST TRANSLATION
Development of genetic tests as an offshoot of research studies
Genetic research studies very often result in information that can be used directly for diagnostic testing in patients and their family members. Taking this into consideration, research studies can incorporate quality control measures at the outset to allow the appropriate use of information generated for diagnostic purposes that have a direct impact on the clinical care of patients and their family members. This was the approach taken for the development of genetic testing for the lissencephaly disorders. From the very beginning, the research laboratory worked in collaboration with the CLIA-certified clinical molecular diagnostic laboratory. All samples from patients enrolled in the research study were received directly in, and processed by, the diagnostic laboratory. DNA was isolated and quantified, and samples were labeled and stored with appropriate documentation of the entire process (identical to that performed for any other sample received in the diagnostic laboratory for genetic testing). This process ensured that the patient samples were handled under quality control guidelines as required by CLIA (1988)1 and the College of American Pathologists (CAP). The diagnostic laboratory stored the patient samples and transferred an aliquot of the DNA to the research laboratory.
The lissencephaly research studies have resulted in the identification of candidate genes (LIS1, DCX, and ARX) and disease-associated mutations.6–8 With the identification of the pathologic mutation in a patient the possibility of genetic testing in other family members became possible. As each mutation was identified in the research laboratory, the diagnostic laboratory confirmed the research result using the aliquot of the patient DNA that had remained in the diagnostic laboratory. Mutation confirmation was performed in all cases by polymerase chain reaction (PCR) and sequence analysis targeted toward the region of the gene containing the mutation identified in the research laboratory. Once the mutations were confirmed by the diagnostic laboratory, the results were reported officially to the patient's physicians. As additional family members requested carrier testing or prenatal diagnosis, this was performed in the diagnostic laboratory on a clinical fee-for-service basis.
The diagnostic laboratory initially performed targeted mutation analysis in the LIS1, DCX, and ARX genes for mutation confirmation in patients followed by additional mutation testing in family members as requested. Once research results were published and the gene and disease correlation well-established and documented, the CLIA-certified clinical molecular diagnostic laboratory set up full gene sequencing tests for all three genes, offering these tests on a clinical, fee-for-service basis. The transition to full gene sequencing in a CLIA-certified diagnostic setting is the ultimate desired outcome as this allows for both negative and positive results to be appropriately reported. Confirmation of positive research findings allows for only positive results to be appropriately reported and is a good intermediate step before full gene analysis is available clinically. Figure 2 depicts the approach toward genetic testing for the lissencephaly genes.

Flowchart depicting the approach taken toward developing genetic testing for lissencephaly disorders.
An important aspect of this approach is the incorporation of the work performed by the diagnostic laboratory, i.e., sample processing, mutation confirmation, and result reporting, into appropriate research funding. The work performed by the diagnostic laboratory was incorporated into the NIH-funded research grant budgets at the outset. Funds were included for a portion of a clinical technologist's salary, clinical laboratory director's salary, and necessary supplies and reagents. The peer reviewers of the grant proposals viewed this research cost very positively, as it ensured the highest level of quality control in sample handling and result reporting to families participating in research. The initial support through research funds was instrumental for the development of lissencephaly testing by the diagnostic laboratory and enabled the subsequent transition of this testing into the clinical arena.
Incorporation of the diagnostic laboratory in the lissencephaly research study allowed for a systematic and quality-controlled approach to the reporting of patient results. This was viewed favorably by both patients and their families, as well as by our institutional review board. A detailed consent form that explained the study, how patient samples would be handled, who had access to patient information, how and when results would be communicated to patients and their physicians, what aspects of the study were performed on a research basis versus clinical (standard of care) basis, and when patients could choose to terminate their participation was presented to patients/family members by an experienced genetic counselor before their inclusion in the research study. This patient consent form was reviewed and approved by the institutional review board before its implementation.
Custom mutation analysis
The underlying principle of this model is the confirmation of research mutation findings in the clinical setting for the use in appropriate clinical management and especially before the testing of other family members or prenatal testing. In this model, the CLIA-certified clinical molecular diagnostic laboratory does not have to be a collaborative partner of the research laboratory, although a close communication between the diagnostic and research laboratories is helpful. This approach was first implemented in the molecular testing for X-linked myotubular myopathy. An investigator had identified mutations in the MTM1 gene in patients with X-linked myotubular myopathy, as part of a research study,9 and required a mechanism for reporting results back to the patients. Confirmation of the presence of the mutation in the patient DNA was made available through the CLIA-certified clinical molecular diagnostic laboratory at the University of Chicago.
An important aspect of mutation confirmation is the requirement of a new blood sample directly from the patient for testing. This is to exclude the possibility of a sample mix up occurring during analysis performed in the research laboratory. For this reason, patient DNA isolated in the research laboratory is not adequate for mutation confirmation testing purposes. In instances where the patient may be deceased and no new blood sample is available for mutation confirmation, the following alternatives can be considered: (i) a newborn blood spot or other sample from the patient that may have been stored in a CLIA/newborn screening laboratory may be used for DNA isolation, (ii) another family member who is an obligate carrier, or known otherwise to be a carrier of the mutation, or another affected family member may be used for mutation confirmation, or (iii) at the very minimum if nothing is available, a DNA sample from the research laboratory needs to be used as a positive control for the assay; however, it is important to indicate in the patient report that no appropriate material was available to clinically confirm the research finding. After mutation confirmation, testing of additional family members for carrier status, presymptomatic diagnosis, or prenatal testing is available. If a mutation is identified in the sample being tested then the research results are confirmed. If a mutation is not present in the sample being tested, however, then the research results cannot be considered confirmed and no further testing should be performed.
Mutation confirmation is usually performed by PCR and sequencing targeted toward the region of the gene containing the mutation previously identified in the research laboratory, i.e., single exon/amplicon PCR and sequencing. The cost of single amplicon PCR and sequencing typically ranges between $200 and $500, and the method is generally easy to set up in a diagnostic laboratory. Mutation confirmation testing is usually performed as a clinical fee-for-service test by the diagnostic laboratory, and testing in all other additional family members once the familial mutation has been confirmed is available as a clinical fee-for-service test. In the instance of MTM1 mutation confirmation in patients with X-linked myotubular myopathy, after mutation confirmation of several patients enrolled in the research study, full gene sequencing of the MTM1 gene was set up in the diagnostic laboratory and offered to patients as a test for mutation identification on a clinical fee-for-service basis. A flowchart outlining the general process of custom mutation analysis is depicted in Figure 3.

Flowchart depicting the approach to custom mutation analysis.
Mutation confirmation and custom mutation analysis can be performed by a CLIA-certified clinical molecular diagnostic laboratory for mutations in virtually any gene. The transition into full gene sequencing for a particular gene, however, will depend on the type of mutations and their distributions in a particular gene, the size of the gene, and thus the resultant cost of setting up full gene sequencing by the diagnostic laboratory.
It is important to note that negative results obtained in a research laboratory, i.e., the absence of a mutation identified in a particular gene, are not amenable to confirmation testing by the diagnostic laboratory with the approaches outlined above. Confirmation would require full gene sequencing and can only be performed at the time such testing is available clinically. Full gene analysis in the diagnostic laboratory is therefore the preferred ultimate outcome.
Some diagnostic laboratories do not offer prenatal testing services, even for those genes for which they provide mutation analysis. Thus, there are times when a second diagnostic laboratory will be called upon to provide prenatal testing for a mutation identified in the primary laboratory. In this case, it is still imperative that the mutation identified in the first laboratory be confirmed in the diagnostic laboratory that is doing the prenatal test, as differences in primers, conditions, or other interlaboratory differences must be shown to have no impact on the likelihood of observing the reported mutation.
COST OF NEW TEST DEVELOPMENT
The cost of new test development will depend largely on the size of the gene to be tested and the technology used for testing. For the majority of cases the technology used is DNA sequencing. The different phases of test development can be broken into the following: preanalytic, analytic, and postanalytic. The preanalytic phase consists mainly of identification of the disease for testing, review of the literature to ensure that adequate information is available to implicate a particular gene with a genetic disorder, retrieval and analysis of gene sequence, and designing of PCR and sequencing primers. Time estimates for performing these activities range from approximately 5 days (for the smaller genes) to 14 days (for the larger genes). The cost associated with this phase of test development has been estimated to be between $1,500 and $3,000. The analytic phase consists of testing, optimizing and streamlining of PCR conditions, sequencing of PCR products, analysis and verification of sequence products, and analysis of nonblinded and blinded samples for validation purposes. This phase of the development can take approximately 6–10 weeks depending on the size of the gene. The cost associated with this phase of the test development can range from $5,000 to $20,000, with the larger estimate being for genes of approximately 30–40 exons. The postanalytic phase consists of documentation of test validation, development of laboratory paperwork, protocols, informational material, uploading information onto laboratory website, and GeneTests as well as informing relevant support groups of the new test availability. The cost associated with this phase of test development has been estimated to be about $1,600. The total cost of development of a new test is roughly estimated to be about $1,000 per exon.
Recovery of test development costs will depend on the price of the test, reimbursement rates and number of tests performed. For example, with a reimbursement rate of approximately 50%, test development costs for the six exon DCX gene (an example of a small size gene) were recovered after performing this test on approximately 60 patients, which took 2–3 years. On the other hand, test development costs for NSD1 (an example of a larger size gene comprised 41 amplicons) were recovered over a one and a half year period and was primarily due to efforts to improve reimbursement rates for this test as well as the higher sample numbers received.
THE IMPORTANCE OF THE GENETIC COUNSELOR
Genetic counselors play a critical, and often multiple, roles in the implementation of testing of ultra rare genetic disorders. Genetic testing requests for ultra rare genetic disorders are usually initiated under two scenarios, (1) when the patient/family comes to the attention of a geneticist at genetics clinic, or (2) when a mutation is identified in a research setting and the research group is looking to confirm their findings in a diagnostic laboratory. In both situations, the genetic counselor can play a crucial role in organizing the provision of appropriate testing. Under the first scenario, it is often the genetic counselor who investigates and communicates with the diagnostic laboratory about the availability of testing. In the case where patients have had a mutation identified through a research study, a genetic counselor can provide the intermediate step and initiate communication between the research and diagnostic laboratories to ensure that the correct information is relayed to allow for accurate test setup. In the second case scenario where the research laboratory initiates the test request, it is often the genetic counselor acting as a research coordinator for the research group, who seeks out the diagnostic laboratory and relays the necessary information for testing purposes. It is therefore important for genetic counselors to understand the nature and different types of testing for ultra rare genetic diseases as well as the role of the research and diagnostic laboratories in the development of ultra rare genetic disease tests (see Scacheri et al., this issue page 337).
CONCLUSION
In this article, two successful models for translating research findings to the diagnostic arena for ultra rare disease genetic testing are described, highlighting key components of partnerships and expertise required. We also describe how the development of new genetic tests can be integrated into research projects and research funding. Guidelines are being developed for the testing of ultra rare genetic disorders and quality control issues are being addressed.10 Experience has shown that genetic testing for ultra rare disorders can be performed in a cost-effective manner and is a viable option for clinical laboratories.
References
Clinical Laboratory Improvement Act, 1988. Available at: http://www.phppo.cdc.gov/clia/regs/toc.aspx. Accessed January 29, 2006.
Andrews LB, Fullarton JE, Holtzman NA, Motulsky AG, editors. Assessing genetic risks: implications for health and social policy. Washington, DC: National Academy Press 1994.
Holtzman NA, Watson MS, editors. Promoting safe and effective genetic testing in the United States—final report of the task force on genetic testing. The National Human Genome Research Institute, Bethesda MD September 1997. Report obtained from website http://www.nhgri.nih.gov/ELSI/TFGT_final/.
Secretary's Advisory Committee on Genetic Testing, 2000. Enhancing the oversight of genetic tests: recommendations of the SACGT. Available at: http://www4.od.nih.gov/oba/sacgt.htm. Accessed January 29, 2006.
Walsh T, Casadei S, Coats KH, Swisher E, et al. Spectrum of mutations in BRCA1, BRCA2, CHEK2 and TP53 in families with high risk of breast cancer. JAMA 2006; 295: 1379–1388.
Lo Nigro C, Chong CS, Smith AC, Dobyns WB, et al. Point mutations and an intragenic deletion in LIS1, the lissencephaly causative gene in isolated lissencephaly sequence and Miller-Dieker syndrome. Hum Mol Genet 1997; 6: 157–164.
Gleeson J, Allen K, Fox J, Lamperti E, et al. Doublecortin, a brain specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell 1998; 92: 63–72.
Kitamura K, Yanazawa M, Sugiyama N, Miura H, et al. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat Genet 2002; 32: 359–369.
de Gouyon BM, Zhao W, Laporte J, Mandel JL, et al. Characterization of mutations in the myotubularin gene in twenty six patients with X-linked myotubular myopathy. Hum Mol Genet 1997; 6: 1499–1504.
Maddalena A, Bale S, Das S, Grody W, et al. Technical standards and guidelines: molecular genetic testing for ultra-rare disorders. Genet Med 2005; 7: 571–583.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: The authors Soma Das and David H. Ledbetter declare no conflict of interest; Sherri J. Bale is a clinical director of GeneDx, Inc.
Rights and permissions
About this article
Cite this article
Das, S., Bale, S. & Ledbetter, D. Molecular genetic testing for ultra rare diseases: models for translation from the research laboratory to the CLIA-certified diagnostic laboratory. Genet Med 10, 332–336 (2008). https://doi.org/10.1097/GIM.0b013e318172838d
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e318172838d
Keywords
This article is cited by
-
Rare disorders have many faces: in silico characterization of rare disorder spectrum
Orphanet Journal of Rare Diseases (2022)
-
Return of Genetic Results in the Familial Dilated Cardiomyopathy Research Project
Journal of Genetic Counseling (2013)
-
The Laboratory‐Clinician Team: A Professional Call to Action to Improve Communication and Collaboration for Optimal Patient Care in Chromosomal Microarray Testing
Journal of Genetic Counseling (2012)
