
Abstract
Purpose: To describe a Gender Assessment Team that has provided a multidisciplinary approach to the diagnosis, medical and surgical treatment, genetic counseling, and psychosocial support of patients with ambiguous genitalia, intersex disorders, and other genital anomalies, collectively termed disorders of sex development; and to determine the major diagnostic categories and approach.
Methods: A retrospective review of 250 patients evaluated by the Team at Children's Hospital and Regional Medical Center in Seattle, WA, from January 1981 through December 2005. The Team included the following specialties: medical genetics, cytogenetics, gynecology, pediatric urology, endocrinology, and psychiatry.
Results: Of the subjects, 177 were infants, 46 were children or adolescents, and 27 had a multisystem genetic condition. The most common diagnoses were congenital adrenal hyperplasia (14%), androgen insensitivity syndrome (10%), mixed gonadal dysgenesis (8%), clitoral/labial anomalies (7%), hypogonadotropic hypogonadism (6%), and 46,XY small-for-gestational-age males with hypospadias (6%).
Conclusion: The six most common diagnoses comprised 50% of the cohort. The expertise of a multidisciplinary team allowed for integrated care for patients with disorders of sex development and identification of novel conditions. Geneticists play an important role in a team approach through knowledge of genetic testing options and diagnosis of patients with karyotypic abnormalities and syndromes with genital anomalies.
Similar content being viewed by others

Main
When a child is born with anomalies of the genitalia, medical providers are faced with the challenge of diagnosing and managing the condition in a timely manner. Although genital anomalies such as cryptorchidism and mild forms of hypospadias and are not uncommon, occurring in up to 2% of newborn males,1 the incidence of actual ambiguous genitalia (where the sex of the child cannot be immediately and confidently established; see Glossary in supplemental materials available online) occurs less frequently. Based on a review of the medical literature by Blackless et al.,1 the frequency of individuals with ambiguous genitalia who may be considered for “corrective” genital surgery is between 1 and 2 per 1000 live births. Examples of ambiguous genitalia include undervirilized XY males (Fig. 1A) and masculinized XX females. Some life-threatening conditions, such as the salt-wasting form of congenital adrenal hyperplasia (CAH), require urgent diagnosis and treatment (Fig. 1B).2,3 Other concerns are psychological ones faced by family members who must make timely decisions about the sex of rearing when genital ambiguity is significant or who must reconcile issues that arise when the appearance of the external genitalia does not match the chromosomal sex. Adolescents may come to medical attention because of genital anomalies or failure to initiate normal pubertal development or menses, and prompt diagnosis and psychosocial support are essential. In the past, the nomenclature used to describe these conditions has included “intersex disorders,” “sex reversal,” “true hermaphrodite,” and “pseudohermaphroditism,” but given the confusion inherent in these terms and their often pejorative connotations, a revised taxonomy has been proposed, with “disorders of sex development” (DSD) as the umbrella term and a specific diagnosis applied when the etiology is known.4–6

Range of appearance of genitalia in infants with disorders of sex development (DSD). (A) 46,XY newborn infant with partial androgen insensitivity syndrome and penoscrotal hypospadias, small phallus with chordee, bilateral cryptorchidism, and absence of müllerian structures on pelvic ultrasonography. The child had a mutation in the androgen receptor gene and was raised as male. (B) 46,XX newborn infant with congenital adrenal hyperplasia and virilized external genitalia with rugation and partial fusion of the labioscrotal folds. No gonads were palpable, and uterus and ovaries were visualized on ultrasound scan. She was raised as female. (C) 46,XY male with penoscrotal hypospadias with transposition. The bifid scrotum extends above the superior portion of the phallus. Bilateral descended gonads were palpable in the scrotum. (D) Infant with ovotesticular DSD at 4 months of age with chimeric 46,XX/46,XY chromosome composition. There were separate vaginal and urethral openings and a palpable left inguinal mass. Bilateral ovotestes were identified. The left-sided gonad composed of predominantly seminiferous tubules and vas deferens was removed; a right-sided hemiuterus, fallopian tube, and gonad with predominantly ovarian tissue were retained. She was assigned female gender.
To address the medical, surgical, and psychosocial challenges posed by such situations, we established a multidisciplinary Gender Assessment Team in 1981 to coordinate the diagnosis, evaluation, and management of patients with DSD. We review our experience over 25 years in treating 250 pediatric patients with more than 25 different conditions affecting genital development. In the glossary (supplemental materials available online), we propose a set of definitions incorporating DSD terminology to facilitate consistent communication about this group of disorders. We highlight the role that geneticists can play in such a multidisciplinary approach.
METHODS
Formation of the Gender Assessment Team
In 1981, clinicians at the University of Washington and Children's Hospital and Regional Medical Center (CHRMC) in Seattle, WA, established a Gender Assessment Team composed of specialists in medical genetics, cytogenetics, gynecology, and reproductive endocrinology and the pediatric specialties of urology, endocrinology, adolescent medicine, and psychiatry. The purpose of the Team has been to provide the following comprehensive care for families and individuals with DSD: diagnostic evaluation using clinical, radiographic, surgical, and laboratory methods, including cytogenetic, endocrinologic, and, more recently, molecular genetic tests; medical and surgical management; genetic counseling for recurrence risks and discussion of etiology and prognosis; and counseling for psychosocial support. The majority of patients have presented in infancy (before 1 year of age), and many newborns were evaluated at the time of admission to CHRMC by several Team members working in a coordinated manner, whereas others were evaluated by one or more Team members on an outpatient basis. The Team also evaluated patients with DSD identified during childhood and adolescence (after 1 year of age), such as those who failed to undergo spontaneous puberty. In general, girls with Turner syndrome, males with Klinefelter syndrome, and many infants with multiple congenital anomalies in whom the diagnosis of a condition with multisystem involvement was established were not referred for full Team evaluation or follow-up, but rather were evaluated by specialists from appropriate disciplines. In many cases, the geneticist who provided the initial evaluation and diagnosis of such infants helped triage their other care needs, which in many cases superseded their genital anomalies. As a result, the Team's experience does not reflect a complete record of all individuals with DSD evaluated by the genetics service at CHRMC. Nonetheless, some patients were referred for Team evaluation and gender assignment before recognition of their more complex medical conditions or syndromes, and a listing of these diagnoses is provided in Table 1 for reference. However, this summary does not attempt to document all the syndromes that can be associated with genital anomalies.
The Team has met every 2 months to discuss the history, diagnosis, and management of new and established patients referred for evaluation. Any Team member can request rediscussion of a patient previously evaluated by the Team. Additional diagnostic tests, treatment options, and recommendations for follow-up for each patient are then discussed with the family by the Team or Team member designated as the contact physician. The long-term follow-up allows for clarification of diagnosis for some individuals and assessment of outcomes for others, although systematic, long-term follow-up has not been pursued, and outcome data are not available for many patients. This review was deemed exempt by the Institutional Review Board at CHRMC.
Gender Assessment Team approach
A general approach has been formulated. In alignment with recommendations of the International Consensus Conference on Intersex Disorders that proposed the DSD terminology,5,6 our Team has consistently promoted the core principles that all individuals must receive a gender assignment after birth that should be based on expert evaluation by a multidisciplinary team. The initial point of contact for the Team is usually the genetic counselor who coordinates the Team and contacts the appropriate specialists, although the on-call endocrinologist or urologist may also receive the initial referral. The first decision point is whether the patient has an isolated genital anomaly or one associated with a multiple congenital anomaly syndrome, such as campomelic dysplasia, Aarskog syndrome, Opitz syndrome, or Prader-Willi syndrome.7,8 Each child with an isolated genital disorder is referred to the Gender Assessment Team, with the goal of establishing the chromosomal sex, the gonadal sex, and the phenotypic sex (appearance of the external and internal genitalia) to determine the underlying diagnosis. The sex of rearing is established for an infant with ambiguous genitalia in the context of a family conference that includes the concerns of the parents and ideally, input from all the Team members. Components of the evaluation include9–11:
-
Maternal history of any symptoms consistent with elevated serum androgen levels or exogenous androgen exposure during pregnancy.
-
Medical history of the use of androgens, growth patterns, and age of menarche for a child or adolescent.
-
Detailed family history of any individuals with infertility, genital abnormalities, delayed puberty, multiple miscarriages, or genital surgery.
-
Physical examination with thorough evaluation of all organ systems and particular attention paid to the external genitalia, including:
-
Measurement of the penis/clitoris. Micropenis is defined as a stretched penile length <2.5 SD below the mean (or <2.0 cm in a term newborn male).12–14 We define clitoromegaly as a clitoris >1 cm in length in a term female.15,16 In infants with extreme ambiguity, differentiation between micropenis and clitoromegaly may be difficult.
-
Determination of the presence of a distinct urethra and vaginal introitus versus a single urogenital sinus.
-
Palpation for gonads in the inguinal canal or labia/scrotum, employing the “soap technique” as necessary (a lubricant such as liquid soap is applied to the inguinal area and the palpating finger is drawn from the pelvic rim toward the labioscrotal fold; often the gonad “slips” from under the finger).17
-
-
Evaluation of the internal genitalia may include:
-
Pelvic ultrasound examination to identify müllerian structures (essential in infants). The fallopian tubes are often not visible by ultrasound evaluation. The uterus is optimally visualized in the first few weeks of life because the maternal estrogen effect thickens the endometrial lining, but pelvic structures can also be identified by ultrasonography at later stages of development.18
-
Laparoscopy and/or laparotomy to elucidate pelvic architecture and gonadal biopsy for histological and/or cytogenetic evaluation. In some cases, ultrasound examination may identify the size and location of gonads, but this is not uniformly reliable.19 The role of pelvic magnetic resonance imaging in identifying internal genital structures remains controversial, particularly for infants, but may be helpful for older children or adolescents.
-
Endoscopy including cystoscopy and colposcopy to define bladder and vaginal anatomy.
-
Fluoroscopic studies such as genitourinary sinography to define anatomic relationships.
-
-
Genetic testing may include the following:
-
Each patient requires a peripheral blood karyotype with evaluation of at least 20 metaphases (and up to 100 if mosaicism is suspected) by routine G-banding techniques at 550 bands or greater. Quinacrine (Q) banding can evaluate the highly variable heterochromatic region of the Y chromosome. In urgent cases, an unstimulated 24- or 48-hour cytogenetic culture may yield preliminary information regarding chromosomal sex; it is followed by a routine 72-hour harvest of a stimulated culture for confirmation of preliminary findings. In general, even if chromosome studies were obtained by amniocentesis, a peripheral blood karyotype is desirable because of increased resolution of chromosome bands and lower likelihood of culture artifact (e.g., maternal cell contamination). Because the leukocytes in blood are a different tissue from the amniocytes used in prenatal diagnosis, analysis of blood represents sampling of a different tissue if mosaicism is suspected.
-
Skin fibroblasts derived from genital skin or other sources may undergo karyotypic analysis.
-
Fluorescence in situ hybridization (FISH) (Fig. 2) to augment routine cytogenetic studies by allowing rapid scanning of metaphase and/or interphase cells for the presence of X and Y chromosome material to evaluate for mosaicism (e.g., 45,X/46,XY) or chimerism (e.g., 46,XX/46,XY) and presence or absence of the sex-determining region on the Y chromosome (SRY).20 Interphase FISH studies may also be performed on thin tissue sections or touch imprints of gonadal biopsies.
Fig. 2 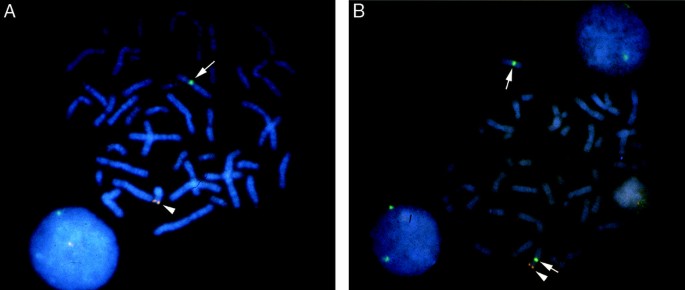
Fluorescence in situ hybridization (FISH) using separate probes for the SRY gene (red; arrowhead) and the X-centromere (green; arrow). (A) Metaphase chromosome spread of a normal 46,XY male showing the green signal at the centromere of the X chromosome (arrow) and red signal at the tip of the short arm of the Y chromosome (arrowhead). (B) Metaphase chromosome spread of a 46,XX male (46,XX testicular DSD) with translocation between the p arms of the X and Y chromosomes, designated t(X;Y). The X chromosome with the translocated portion of Yp has both the red SRY gene signal (arrowhead) and green X centromere signal (arrow). The normal X chromosome is indicated by the white arrow only. Chromosomes are counterstained blue with DAPI to allow visualization.
-
Specialized molecular genetic testing for Y chromosome deletions that may be associated with infertility21 if the Y appears abnormal.
-
Other specialized genetic tests: for example, molecular genetic testing for mutations in the CYP21A2 gene for patients with CAH and suspected 21-hydroxylase deficiency (21-OHD), in the androgen receptor gene (AR) for patients with suspected androgen insensitivity syndrome,22 in the SRY gene for patients with XY gonadal dysgenesis,23 in the SOX9 gene in campomelic dysplasia,24,25 or in the WT1 gene in Frasier and Denys-Drash syndromes.26 The list of genes known to be involved in DSD continues to expand.5,6
-
-
Endocrinologic evaluation is individualized, but usually involves:
-
Assessment of the function of the hypothalamic/pituitary/gonadal axis. Studies routinely include a gonadotropin profile (serum concentrations of luteinizing hormone [LH] and/or follicle-stimulating hormone [FSH]); assessment of gonadal function (serum concentrations of testosterone and/or estradiol); and assessment of other pituitary axes when indicated. These tests are most revealing when obtained between 2 and 4 months of life when infants normally undergo a “mini-pubertal” increase in LH, FSH, testosterone, and estrogen.27
-
Hormonal stimulation tests such gonadotropin-releasing hormone (Lupron) or human chorionic gonadotropin may be performed to determine the ability of the pituitary and gonads, respectively, to respond to upstream regulatory hormones.
-
Adrenal hormone production may be assessed by measurement of the serum concentrations of sodium, potassium, and 17-hydroxyprogesterone, particularly in infants presenting with CAH. Abnormal serum concentrations of other adrenal hormones, such as dihydroepiandrosterone, androstenedione, cortisol, and steroid hormone intermediates, may indicate a block in the testosterone biosynthetic pathway.11
-
Measurement of testosterone/dihydrotestosterone ratio to evaluate suspected 5-α-reductase deficiency.28
-
Measurement of müllerian-inhibiting substance (MIS) and/or inhibin B to assess testicular function in children with nonpalpable gonads.29
-
-
Psychological evaluation to identify the concerns of the parents of a newborn with ambiguous genitalia and provide support for the accompanying uncertainty.30 This evaluation includes an assessment of parents' expectations regarding the gender of their baby, their understanding of what they have been told since the baby's birth, and their preferences, particularly within the context of their cultural and/or religious background, as well as a general assessment of coping strategies. Decisions regarding medical interventions and sex of rearing incorporate the wishes of family members using a collaborative model. Ongoing psychological support for children and adolescents with genital anomalies and their parents is provided by the psychologist (E. McCauley) who has more than 25 years of experience in treating individuals with DSD. This emphasis on open communication, participation of family members and/or patients in the decision-making process, and maintenance of strict confidentiality are core principles for optimal management as set forth in the consensus statement regarding management of individuals with DSD.5,6

RESULTS
The 250 patients evaluated by the Gender Assessment Team are summarized in Table 1.
Infancy
A total of 177 (71%) presented in infancy. Of these, the majority had abnormal genitalia with Y chromosome material present (98/177; 55%), and 71 of 177 (40%) had the 46,XX female karyotype (Table 2). In eight of 177 infants (5%), the appearance of the external genitalia did not match the chromosomal sex, which we have labeled chromosome-phenotype discordance; these infants had normal genitalia at birth, although the prenatal karyotype was either mosaic or did not match the ultrasound appearance of the genitalia. Although the focus of the Team has been on infants and children with disorders restricted to the genitalia, 27 patients with 22 different multisystem genetic syndromes were discussed (Table 1), including three infants with cloacal exstrophy and three with panhypopituitarism. One infant had X-linked lissencephaly and agenesis of the corpus callosum with ambiguous genitalia (XLAG),31 a disorder caused by mutations in the ARX gene.32 Overall, we failed to establish a diagnosis for 29 individuals, representing 12% of the cohort.
The 98 infants with Y chromosome material have been subdivided into those with hypospadias with or without micropenis (61%), those with isolated micropenis (38%), and one with aphallia (1%) (Table 2). The largest group of infants with hypospadias had mixed gonadal dysgenesis33; several had Y chromosome abnormalities (e.g., pseudodicentric Y with or without mosaic loss of the Y chromosome34), and the gender assignment was based on the degree of masculinization and/or parental choice. Fifteen were 46,XY males with hypospadias who were markedly small for gestational age, an association that has been previously reported.35,36 Eleven exhibited a distinctive genital configuration described as penoscrotal hypospadias with transposition and were raised as male (Fig. 1C).37 Three had both ovarian and testicular tissue and at least one XY cell line (Fig. 1D); two were 46,XX males (now termed 46,XX testicular DSD) and had Y chromosome material including SRY identifiable by FISH analysis (Fig. 2).38,39
In the infants with Y chromosome material and micropenis without hypospadias, there was a range of disorders (Table 2), including three patients with an unusual gonadotropin profile previously reported that may represent a new syndrome.40 Assigned sex for these infants varied and was based on parental preferences and the best estimate of future genitourinary function, sexual function, and fertility.
The one patient with aphallia presented with bilaterally descended testes and was raised as female after orchiectomy.41
The diagnoses for the 71 infants with a 46,XX karyotype included (1): CAH caused by 21-hydroxylase deficiency (49%); (2) clitoral and/or labial abnormalities (24%); (3) partially masculinized females (46,XX karyotype, normal ovaries) who had cloacal abnormalities or a urogenital sinus (10%), some of whom fall within the VATER/VACTERL association spectrum (vertebral anomalies, anal atresia, cardiac malformations, tracheoesophageal fistula, renal anomalies, and radial ray limb defects)42; (4) vaginal anomalies (8%); and (5) those with 46,XX testicular DSD who were SRY negative by FISH analysis (six infants, with four assigned as male and two assigned as female). With the exception of the most severely virilized females with CAH and those with 46,XX testicular DSD, almost all had a predominantly female phenotype. All 46,XX individuals with CAH were raised as female, an assignment supported in previous series and the consensus statement.5,6
The diagnoses in the eight infants presenting with chromosome-phenotype discordance but normal-appearing genitalia included those with a 45,X/46,XY mosaic karyotype detected prenatally by amniocentesis performed for advanced maternal age; at birth, all had normally masculinized external genitalia. It is likely that these phenotypic males would not have come to medical attention if not for the prenatal chromosome studies.43 Two infants had a prenatal ultrasound genitalia appearance seemingly at odds with the karyotype obtained by amniocentesis; one was the result of an amniocentesis sample mix up, and the other had a buried penis but otherwise normal male appearance at birth.
Childhood/adolescence
In the 46 patients with childhood or adolescent presentation (Table 3), the following observations were made: (1) Thirty-nine percent (18/46) had chromosome-phenotype discordance disorders characterized by a 46,XY karyotype with female external genitalia, of which the majority had complete androgen insensitivity syndrome (CAIS). Typical presentation in childhood was a phenotypically normal female with inguinal hernia(s) containing testes; typical presentation in adolescence was a phenotypically normal female with absent adrenarche and primary amenorrhea, absent müllerian structures on ultrasound examination, and high serum concentrations of testosterone and LH. (2) Males with isolated micropenis were most likely to have hypogonadotropic hypogonadism (22%), with low serum concentrations of FSH and LH. Two patients with anosmia were diagnosed with Kallmann syndrome. (3) Several females had adolescent presentation of vaginal or uterine anomalies within the spectrum of Mayer-Rokitansky-Kuster-Hauser syndrome.44,45 (4) Six had pure gonadal dysgenesis with a spectrum of genital anomalies and karyotypes. (5) Two had primary ovarian failure of unclear etiology with a normal 46,XX karyotype and female phenotype.
DISCUSSION
Most common diagnoses
Fifty percent of the patients discussed by the Team had one of six diagnoses (Table 4). (1) The most common condition was CAH due to 21-OHD, a finding consistent with its birth prevalence of 1:15,000.3,46 Most pediatricians are familiar with 21-OHD, as affected female infants are virilized at birth, the majority have salt-wasting with hyponatremia and hyperkalemia, and they may present in adrenal crisis.2 We routinely test for the common mutations in the CYP21A2 gene in these infants, with full gene sequencing if necessary, to confirm the diagnosis, provide information on genotype-phenotype correlations, and allow for prenatal diagnosis and management in future pregnancies. (2) The second most common diagnosis was androgen insensitivity syndrome, either partial (PAIS) or complete (CAIS). In contrast to newborns with PAIS who may have undervirilized external genitalia at birth of varying severity,47 those with CAIS have normal female external genitalia. The diagnosis of CAIS may be suspected in a newborn or adult with a family history of this X-linked disorder; sequencing of the AR gene encoding the androgen receptor detects mutations in >95% of those with CAIS22 and is standard practice for our patients with this diagnosis, particularly given the importance of this information for decision making regarding recurrence risks among family members. The diagnosis of PAIS remains challenging in infancy as the yield for AR sequencing is unknown and gonadotropin concentrations can be variable, even paradoxical.48 In our experience, the identification of a causative AR mutation in this population is rare, although we often perform this testing. (3) In this series, approximately 50% of males with mixed gonadal dysgenesis had 45,X/46,XY mosaicism, a useful diagnostic clue; in contrast, the diagnosis of mixed gonadal dysgenesis in those with a normal 46,XY karyotype is often not established until the time of laparoscopy with gonadal biopsy. (4) Seven percent had labial and/or clitoral anomalies. (5) Male children with micropenis without hypospadias are most likely to have hypogonadotropic hypogonadism identified when they fail to enter puberty, although some are also diagnosed in infancy during evaluation of micropenis; however, interpretation of serum concentrations of LH and FSH can be difficult after the mini-pubertal surge (between 2 and 4 months) and before adolescence. (6) A relatively common condition was hypospadias with or without cryptorchidism in males with a normal 46,XY karyotype and intrauterine growth restriction who are often born prematurely.35,36 In this group, male sex of rearing appears to be appropriate, although limited long-term data are available regarding functional outcomes, and the etiology is unknown.
Comparison with other reported series
This experience with 250 different patients with anomalous genitalia or DSD over 25 years represented approximately 10 new patient evaluations each year or seven to eight infants presenting with DSD each year. Few other series of this scope have been reported in the medical literature. Similar to our results, one series of 51 children ascertained over a 17-year period in Australia49 identified 21-OHD as the most common diagnosis, representing 73% of 46,XX patients and 31% of the entire cohort. This group also reported a wider range of diagnoses for those with 46,XY karyotype; 17% of this group had androgen insensitivity syndrome, and 17% had gonadal dysgenesis (either pure or mixed). In that series, almost 25% had no final diagnosis. Our cohort comprised a larger variety of specific diagnoses, particularly those based on chromosomal anomalies, perhaps reflecting ascertainment bias because of the large number of geneticists who actively refer patients to the Team. In another survey of 291 infants with ambiguous genitalia, mostly composed of undervirilized males, a diagnosis of PAIS was made in 145 (50%) newborns and dysgenetic testes were identified in an additional 11%.50 However, the purpose of that survey was to develop a masculinization scoring method for newborns, and the diagnostic categories were not well defined. The diagnosis of PAIS is problematic given the variable definition of the term “partial” across series. In another review of 167 patients, the most common diagnoses in infancy were mixed gonadal dysgenesis (13%) and CAH (9%), with disorders such as androgen insensitivity syndrome (11%) more common in older children and adolescents.51 That group identified 14 (8%) with 46,XY pure gonadal dysgenesis, in contrast to only six reported here (2%).
Recognition of novel groups of patients
One of the most valuable aspects of the multidisciplinary approach has been the recognition of novel associations involving genital anomalies, including conditions underreported in the medical literature, such as small-for-gestational-age males with hypospadias and a normal 46,XY karyotype. These males typically have no other malformations. The knowledge of this association provides reassurance that male sex assignment is appropriate and routine management of hypospadias is in order. Another condition is that of otherwise normal infants identified prenatally with 45,X/46,XY mosaicism, often by amniocentesis for advanced maternal age, who most often will have a normal male phenotype at birth. However, because few of these males with mosaicism have reached adulthood, their risk of short stature and infertility is unknown. This karyotype can also be observed in girls with features of Turner syndrome, with or without ambiguous genitalia, or males with mixed gonadal dysgenesis.34 A third condition is that of 46,XY males with hypospadias and penoscrotal transposition. In the absence of cryptorchidism or other congenital anomalies, these males appear to have normal male gonadal function (D. F. Gunther, MD, Children's Hospital, Seattle, WA, personal observation), although fertility remains to be established.
Our group has also identified at least two new syndromes: (1) three males (including two brothers) with micropenis without hypospadias, testicular regression with poor response to HCG stimulation, and microcephaly40 and (2) a phenotypic female with XLAG syndrome (X-linked lissencephaly with ambiguous genitalia) and 46,XY karyotype, one of the first to be described.31,32
Evolution of the approach
Certain themes have emerged as the Team reviewed its experience and tried to respond to the recommendations of the consensus statement.5,6 The sophistication of cytogenetic, molecular, and endocrinologic testing options has increased over the past two decades. Improved chromosome banding techniques have allowed the identification of subtle chromosome differences, and FISH technology has facilitated rapid detection of X and Y chromosomes and, hence, identification of chromosomal mosaicism. The clinical availability of molecular sequencing of the AR gene has simplified the diagnosis of CAIS, which formerly depended on labor-intensive, insensitive androgen-binding assays of genital skin fibroblasts.52 Molecular genetic testing has also facilitated diagnosis of a wide variety of other genetic disorders.5,6 New hormone assays continue to be developed.
The surgical approaches to treatment have also evolved. Because of concerns raised by patient advocacy groups, the publication of sensational cases in the press, and discussion raised by the consensus statement, the appropriateness of genital surgery in infancy has been more widely questioned by patients and physicians. In most cases, it is now our policy to present surgical correction of the genitalia as an option, with the understanding that few objective data exist as to which approach is more desirable (surgery in infancy or delaying until later in life when the individual can participate in the decision-making process). Families are provided with information about the medical, functional, and cosmetic rationale for urologic and/or genital surgery. For example, removal of asymptomatic testes in females with CAIS is now more likely to be deferred in childhood than in the past to allow the benefit of hormonal production at the time of puberty; however, the consensus statement suggestion to retain these gonads throughout adulthood given the relatively low risk of germ cell malignancy5,6 has not yet been adopted by our Team. In practice, the decision to defer surgery has typically occurred in cases such as a clitoral reduction surgery for mildly virilized females with CAH, a trend noted in the consensus statement,5,6 but not when more involved surgical corrections were chosen (such as perineal hypospadias and cryptorchidism repair for a 46,XY infant with a DSD). Generally, our experience has been that most parents still choose to proceed with genitoplasty in infancy; however, some parents in the past few years have elected to postpone surgery until later in life.
In recent years, the Team, in consultation with parents, has recommended reconstruction of infants with 46,XY DSD as male when practical, whereas in the past, some centers might have suggested assigning and raising such a child as female if surgical intervention was deemed too complex. Because long-term follow-up studies are lacking, weighing the risks and benefits of a specific gender assignment in these situations is difficult. Decisions regarding the appropriate sex-of-rearing for infants with significantly ambiguous genitalia are often challenging.53,54 In one study on satisfaction with gender assignment in 46,XY adults born with perineoscrotal hypospadias, at least 76% of participants were satisfied with their assigned sex, whether male or female, although almost 50% were dissatisfied with their body image, and two thirds had some degree of dissatisfaction with their sexual function.55 Studies of girls with CAH have established that prenatal androgen exposure is associated with increased frequency of cross-gender role behavior, but effects on gender identity are less evident.56 Some surveys suggest that prenatal androgen exposure may play a more central role in gender identity development, particularly if a Y chromosome is present.57,58 In one cohort of 84 subjects with DSD who were 5 years of age or older, 53% of those with a Y chromosome and reared as female later declared themselves as male.59 In another study, 13% of the 54 children raised as female and none of the five children raised as male reported gender identity confusion, whereas 46% of the girls had “boyish” conduct.60 Factors associated with gender identity confusion included pre- and postnatal androgenization of the brain, cross-gender role behaviors, parental inability to accept the sex assignment, repeated alteration of sex assignment, and family problems.60
Because of the complex issues involved, some general guidelines for determination of sex assignment have been developed.5,53,54 A theme of management for our patients has been to match sex-of-rearing with chromosomal and gonadal sex whenever possible, with the recognition that prenatal hormone exposure influences gender role behavior, whereas gender identity can develop as male or female over wide variations in gender roles.61 In addition, our practice includes a large educational component, providing resources from information-based Web sites (such as http://www.sickkids.ca/childphysiology/cpwp/Genital/genitaldevelopment.htm and http://www.dsdguidelines.org/) and patient education/advocacy groups such as the Intersex Society of North America (www.isna.org). We acknowledge the controversies surrounding surgical interventions so an informed decision can be made.62,63 Psychosocial support for families faced with a decision about gender assignment is a priority, and parents are given pragmatic, age-appropriate recommendations for disclosure of a diagnosis of a DSD to a child in an honest, nonstigmatizing manner. This approach is supported by the results from an Australian multidisciplinary team that applied a patient-centered, holistic model of care: overall, despite some dissatisfaction with sexual activity and function, the adults had mostly positive psychosocial outcomes.58 Nonetheless, despite our attempts to provide appropriate long-term follow-up and psychological support as well as facilitate transition of our pediatric patients with DSD to adult care, the Team has not always succeeded in fulfilling this goal.5,6 We are exploring other medical models, including involvement of social work and other support services, and regularly scheduled DSD Clinic dates to better coordinate longitudinal care and facilitate measurable outcome studies. Because the number of objective studies published on the correlation between gender identity, psychosocial outcomes, and timing of surgery is limited, we hope that the data collected by our Team and other multispecialty clinics will provide guidance for best future practices.
Potential value of the multidisciplinary team approach
We believe that a multidisciplinary team approach to patients with DSD is effective because each specialty can focus on its strengths in concert with other specialists. The camaraderie, trust, and comfort level of the Team, which have evolved though the collective experience of the group (many of whom have been members since its inception 25 years ago), create a level of expertise that can provide the most comprehensive care possible for these patients. Although endocrinologists have often taken the lead in evaluating infants with DSD,5,6 the importance of geneticists as integral members of a team is underscored by the large numbers of patients with karyotypic anomalies, especially involving the sex chromosomes, the challenges in interpreting these findings, and the desire for recurrence risk information by families. When a team approach is successful, the timely diagnosis, management, and psychosocial support for patients and/or parents provide reassurance to families seeking immediate and comprehensible solutions for these medically and socially complex problems. The Team often serves as a regional referral center because its experience has often proved invaluable in developing diagnostic and treatment plans for patients with rare conditions and those for whom initial management was incomplete or suboptimal.
Future directions
For many of the established diagnostic categories, the molecular basis of the disorders remains unknown. However, as an understanding of the genes involved in developmental pathways emerges, improved molecular tests will facilitate specific diagnoses for patients with DSD, and the role of the geneticist in coordinating appropriate genetic testing and providing recurrence risk information will continue to expand. In the future, a partnership between basic scientists who study sexual differentiation, clinicians from multiple disciplines with expertise in childhood gender assessment, parents of children with genital differences, and patient advocacy groups will be most effective in providing the optimal care for infants and children with anomalous genitalia and DSD.
References
Blackless M, Charuvastra A, Derryck A, Fausto-Sterling A, et al. How sexually dimorphic are we? Review and synthesis. Am J Human Biol 2000; 12: 151–166.
Clayton PE, Miller WL, Oberfield SE, Ritzen EM, et al. Consensus statement on 21-hydroxylase deficiency from the European Society for Paediatric Endocrinology and the Lawson Wilkins Pediatric Endocrine Society. Horm Res 2002; 58: 188–195.
Pang SY, Wallace MA, Hofman L, Thuline HC, et al. Worldwide experience in newborn screening for classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Pediatrics 1988; 81: 866–874.
Dreger AD, Chase C, Sousa A, Gruppuso PA, et al. Changing the nomenclature/taxonomy for intersex: a scientific and clinical rationale. J Pediatr Endocrinol Metab 2005; 18: 729–733.
Hughes IA, Houk C, Ahmed SF, Lee PA . Consensus statement on management of intersex disorders. Arch Dis Child 2006; 91: 554–563.
Lee PA, Houk CP, Ahmed SF, Hughes IA . Consensus statement on management of intersex disorders. International Consensus Conference on Intersex. Pediatrics 2006; 118: e488–500.
McGillivray BC . The newborn with ambiguous genitalia. Semin Perinatol 1992; 16: 365–368.
Pagon RA . Diagnostic approach to the newborn with ambiguous genitalia. Pediatr Clin North Am 1987; 34: 1019–1031.
Evaluation of the newborn with developmental anomalies of the external genitalia. American Academy of Pediatrics. Committee on Genetics. Pediatrics 2000; 106: 138–142.
Aaronson IA . The investigation and management of the infant with ambiguous genitalia: a surgeon's perspective. Curr Probl Pediatr 2001; 31: 168–194.
Grumbach MM, Hughes IA, Conte FA . Disorders of sex differentiation. In: Larsen PR, Kronenberg HM, Melmed S, Polonsky KS, editors, Williams textbook of endocrinology, 10th ed. Philadelphia: WB Saunders, 2003; 842–1002.
Feldman KW, Smith DW . Fetal phallic growth and penile standards for newborn male infants. J Pediatr 1975; 86: 395–398.
Lee PA, Mazur T, Danish R, Amrhein J, et al. Micropenis. I. Criteria, etiologies and classification. Johns Hopkins Med J 1980; 146: 156–163.
Schonfeld WA, Beebe GW . Normal growth and variation in the male genitalia from birth to maturity. J Urol 1942; 48: 759–777.
Oberfield SE, Mondok A, Shahrivar F, Klein JF, et al. Clitoral size in full-term infants. Am J Perinatol 1989; 6: 453–454.
Phillip M, De Boer C, Pilpel D, Karplus M, et al. Clitoral and penile sizes of full term newborns in two different ethnic groups. J Pediatr Endocrinol Metab 1996; 9: 175–179.
Skoog S . Office pediatric urology. In: Gillenwater JY, Howards F, Mitchell M, editors. Adult and pediatric urology, 4th ed. Philadelphia: Lippincott, Williams, and Wilkins, 2002; 2689.
Garel L, Dubois J, Grignon A, Filiatrault D, et al. US of the pediatric female pelvis: a clinical perspective. Radiographics 2001; 21: 1393–1407.
Herter LD, Golendziner E, Flores JA, Becker E Jr, et al. Ovarian and uterine sonography in healthy girls between 1 and 13 years old: correlation of findings with age and pubertal status. AJR Am J Roentgenol 2002; 178: 1531–1536.
Sinclair AH, Berta P, Palmer MS, Hawkins JR, et al. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature 1990; 346: 240–244.
Reijo R, Lee TY, Salo P, Alagappan R, et al. Diverse spermatogenic defects in humans caused by Y chromosome deletions encompassing a novel RNA-binding protein gene. Nat Genet 1995; 10: 383–393.
Gottlieb B, Beitel LK, Trifiro MA . Androgen insensitivity syndrome. GeneReviews at GeneTests-GeneClinics: Medical Genetics Information Resource (database online) (April 8, 2004). Available at: http://www.geneclinics.org or http://www.genetests.org. Accessed November 14, 2004.
Hawkins JR . Mutational analysis of SRY in XY females. Hum Mutat 1993; 2: 347–350.
Meyer J, Sudbeck P, Held M, Wagner T, et al. Mutational analysis of the SOX9 gene in campomelic dysplasia and autosomal sex reversal: lack of genotype/phenotype correlations. Hum Mol Genet 1997; 6: 91–98.
Foster JW, Dominguez-Steglich MA, Guioli S, Kowk G, et al. Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene. Nature 1994; 372: 525–530.
Mueller RF . The Denys-Drash syndrome. J Med Genet 1994; 31: 471–477.
Grumbach MM, Styne DM . Puberty: ontogeny, neuroendocrinology, physiology, and disorders. In: Larsen PR, Kronenberg HM, Melmed S, Polonsky KS, editors, Williams textbook of endocrinology, 10th ed. Philadelphia: WB Saunders, 2003; 1115–1286.
Sinnecker GH, Hiort O, Dibbelt L, Albers N, et al. Phenotypic classification of male pseudohermaphroditism due to steroid 5 alpha-reductase 2 deficiency. Am J Med Genet 1996; 63: 223–230.
Lee MM, Donahoe PK, Silverman BL, Hasegawa T, et al. Measurements of serum mullerian inhibiting substance in the evaluation of children with nonpalpable gonads. N Engl J Med 1997; 336: 1480–1486.
Berenbaum SA . Management of children with intersex conditions: psychological and methodological perspectives. Growth Genet Horm 2003; 19: 1–6.
Sybert VP, Pagon RA, Ramsdell L, Marymee K . Re: true agonadism: report of a case analyzed with Y-specific DNA probes. Am J Med Genet 1995; 55: 113.
Kitamura K, Yanazawa M, Sugiyama N, Miura H, et al. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat Genet 2002; 32: 359–369.
Mendez JP, Ulloa-Aguirre A, Kofman-Alfaro S, Mutchinick O, et al. Mixed gonadal dysgenesis: clinical, cytogenetic, endocrinological, and histopathological findings in 16 patients. Am J Med Genet 1993; 46: 263–267.
Hsu LY . Phenotype/karyotype correlations of Y chromosome aneuploidy with emphasis on structural aberrations in postnatally diagnosed cases. Am J Med Genet 1994; 53: 108–140.
Gatti JM, Kirsch AJ, Troyer WA, Perez-Brayfield MR, et al. Increased incidence of hypospadias in small-for-gestational age infants in a neonatal intensive-care unit. BJU Int 2001; 87: 548–550.
Hussain N, Chaghtai A, Herndon CD, Herson VC, et al. Hypospadias and early gestation growth restriction in infants. Pediatrics 2002; 109: 473–478.
Pinke LA, Rathbun SR, Husmann DA, Kramer SA . Penoscrotal transposition: review of 53 patients. J Urol 2001; 166: 1865–1868.
Margarit E, Soler A, Carrio A, Oliva R, et al. Molecular, cytogenetic, and clinical characterisation of six XX males including one prenatal diagnosis. J Med Genet 1998; 35: 727–730.
Vilain EJ . 46,XX testicular disorder of sex development. GeneReviews at GeneTests-GeneClinics: Medical Genetics Information Resource (database online) (April 5, 2006). Available at: http://www.geneclinics.org or http://www.genetests.org. Accessed April 22, 2006.
Parisi MA, Kletter GB, Grady R, Mitchell M, et al. Micropenis with testicular regression, low LH levels, and poor androgen and HCG responses: a distinct syndrome?. Am J Med Genet 2002; 109: 271–277.
Skoog SJ, Belman AB . Aphallia: its classification and management. J Urol 1989; 141: 589–592.
Wenstrup RJ, Pagon RA . Female pseudohermaphroditism with anorectal, mullerian duct, and urinary tract malformations: report of four cases. J Pediatr 1985; 107: 751–754.
Telvi L, Lebbar A, Del Pino O, Barbet JP, et al. 45,X/46,XY mosaicism: report of 27 cases. Pediatrics 1999; 104: 304–308.
Spence JE . Vaginal and uterine anomalies in the pediatric and adolescent patient. J Pediatr Adolesc Gynecol 1998; 11: 3–11.
Strubbe EH, Cremers CW, Willemsen WN, Rolland R, et al. The Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome without and with associated features: two separate entities?. Clin Dysmorphol 1994; 3: 192–199.
New MI, Betensky B . Congenital adrenal hyperplasia. GeneReviews at GeneTests-GeneClinics: Medical Genetics Information Resource (database online) (April 15, 2004). Available at: http://www.geneclinics.org or http://www.genetests.org. Accessed November 22, 2004.
Sinnecker GHG, Hiort O, Nitsche EM, Holterhus P-M, et al. Functional assessment and clinical classification of androgen sensitivity in patients with mutations of the androgen receptor gene. Eur J Pediatr 1997; 156: 7–14.
Bouvattier C, Carel JC, Lecointre C, David A, et al. Postnatal changes of T, LH, and FSH in 46,XY infants with mutations in the AR gene. J Clin Endocrinol Metab 2002; 87: 29–32.
Al-Agha AE, Thomsett MJ, Batch JA . The child of uncertain sex: 17 years of experience. J Paediatr Child Health 2001; 37: 348–351.
Ahmed SF, Khwaja O, Hughes IA . The role of a clinical score in the assessment of ambiguous genitalia. BJU Int 2000; 85: 120–124.
Pantzar T, McGillivray BC . A gender assessment team: the British Columbia experience 1982–1996. Am J Hum Genet 1996; 59: A338.
Nitsche EM, Hiort O . The molecular basis of androgen insensitivity. Horm Res 2000; 54: 327–333.
Diamond M, Sigmundson HK . Management of intersexuality. Guidelines for dealing with persons with ambiguous genitalia. Arch Pediatr Adolesc Med 1997; 151: 1046–1050.
Reiner WG . Sex assignment in the neonate with intersex or inadequate genitalia. Arch Pediatr Adolesc Med 1997; 151: 1044–1045.
Migeon CJ, Wisniewski AB, Gearhart JP, Meyer-Bahlburg HF, et al. Ambiguous genitalia with perineoscrotal hypospadias in 46,XY individuals: long-term medical, surgical, and psychosexual outcome. Pediatrics 2002; 110: e31.
Berenbaum SA, Bailey JM . Effects on gender identity of prenatal androgens and genital appearance: evidence from girls with congenital adrenal hyperplasia. J Clin Endocrinol Metab 2003; 88: 1102–1106.
Meyer-Bahlburg HF . Gender identity outcome in female-raised 46,XY persons with penile agenesis, cloacal exstrophy of the bladder, or penile ablation. Arch Sex Behav 2005; 34: 423–438.
Warne G, Grover S, Hutson J, Sinclair A, et al. A long-term outcome study of intersex conditions. J Pediatr Endocrinol Metab 2005; 18: 555–567.
Reiner WG . Gender identity and sex-of-rearing in children with disorders of sexual differentiation. J Pediatr Endocrinol Metab 2005; 18: 549–553.
Slijper FM, Drop SL, Molenaar JC, de Muinck Keizer-Schrama SM, et al. Long-term psychological evaluation of intersex children. Arch Sex Behav 1998; 27: 125–144.
Meyer-Bahlburg HF . Gender assignment and reassignment in 46,XY pseudohermaphroditism and related conditions. J Clin Endocrinol Metab 1999; 84: 3455–3458.
Eugster EA . Reality vs recommendations in the care of infants with intersex conditions. Arch Pediatr Adolesc Med 2004; 158: 428–429.
Frader J, Alderson P, Asch A, Aspinall C, et al. Health care professionals and intersex conditions. Arch Pediatr Adolesc Med 2004; 158: 426–428.
Acknowledgements
The authors acknowledge the contributions of past and present members of the Gender Assessment Team, including the following individuals: Beth Andersen, Carol Bach, Neil Caplin, Mary Beth Dinulos, Christine Disteche, Victor Fujimoto, Ann Giesel, Ian A. Glass, Mark C. Hannibal, Anne Hing, Louanne Hudgins, Byron Joyner, Dana Knutzen, Thomas Lendvay, Kathleen A. Leppig, Cathy Lum, C. Patrick Mahoney, Maila Penttinen, Michael Raff, Meera Ramayya, Penny Schubert, Darci L. Sternen, and Karen Tsuchiya. They especially thank the patients and their families who have been faced with challenging conditions and who have provided motivation and inspiration for our group.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: The authors declare no conflict of interest.
A supplementary glossary is available via the ArticlePlus feature at www.geneticsinmedicine.org. Please go to the June issue and click on the ArticlePlus link posted with the article in the Table of Contents to view this material.
Rights and permissions
About this article
Cite this article
Parisi, M., Ramsdell, L., Burns, M. et al. A Gender Assessment Team: experience with 250 patients over a period of 25 years. Genet Med 9, 348–357 (2007). https://doi.org/10.1097/GIM.0b013e3180653c47
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e3180653c47
Keywords
This article is cited by
-
Discordant sex between fetal screening and postnatal phenotype requires evaluation
Journal of Perinatology (2019)
-
Disorders of sex development in a developing country: perspectives and outcome of surgical management of 39 cases
Pediatric Surgery International (2015)
-
Advances in diagnosis and care of persons with DSD over the last decade
International Journal of Pediatric Endocrinology (2014)
-
Management of children with disorders of sex development: 20-year experience in southern Thailand
World Journal of Pediatrics (2014)
-
Practical approach to steroid 5alpha-reductase type 2 deficiency
European Journal of Pediatrics (2011)
