
Abstract
Several constitutional rearrangements, including deletions, duplications, and translocations, are associated with 22q11.2. These rearrangements give rise to a variety of genomic disorders, including DiGeorge, velocardiofacial, and conotruncal anomaly face syndromes (DGS/VCFS/CAFS), cat eye syndrome (CES), and the supernumerary der(22)t(11;22) syndrome associated with the recurrent t(11;22). Chromosome 22-specific duplications or low copy repeats (LCRs) have been directly implicated in the chromosomal rearrangements associated with 22q11.2. Extensive sequence analysis of the different copies of 22q11 LCRs suggests a complex organization. Examination of their evolutionary origin suggests that the duplications in 22q11.2 may predate the divergence of New World monkeys 40 million years ago. Based on the current data, a number of models are proposed to explain the LCR-mediated constitutional rearrangements of 22q11.2.
Similar content being viewed by others


Main
Although chromosome 22 represents only 2% of the haploid human genome,1 recurrent, clinically significant, acquired, and somatic rearrangements of this chromosome are associated with multiple malignant diseases and developmental abnormalities (reviewed by Kaplan et al.2).The majority of these recurrent rearrangements take place within 22q11.2, suggesting genomic instability related to the structure of this region of human chromosome 22.
The nonrandom chromosome 22 abnormalities include acquired tumor-associated rearrangements such as the t(9;22) associated with acute lymphocytic leukemia (ALL) and chronic myeloid leukemia (CML), the t(8;22) variant translocation associated with Burkitt’s lymphoma, and the t(11;22) of Ewing’s sarcoma (ES) and peripheral neuroepithelioma (NE). The recurrent constitutional abnormalities of 22q include the duplications associated with the supernumerary bisatellited marker chromosome of cat eye syndrome (CES),3 the translocations which give rise to the recurrent t(11;22) malsegregation-derived supernumerary der(22)t(11;22) syndrome,4–6 and the translocations and deletions associated with DiGeorge, velocardiofacial, and conotruncal anomaly face syndromes (DGS/VCFS/CAFS).7–14
The 22q11.2 deletion syndrome, which includes DGS/VCFS/CAFS, is the most common microdeletion syndrome. The overwhelming majority of deleted patients share a common 3 Mb hemizygous deletion of 22q11.2. The remaining patients include those who have smaller deletions nested within the 3 Mb typically deleted region (TDR) and several individuals with rare deletions that have no overlap with the TDR (reference 15 and references within). The entire 3 Mb TDR has recently been sequenced, permitting detailed examination of this region of chromosome 22. Four copies of chromosome 22-specific duplicated sequence or low copy repeats (LCRs) within the 3 Mb TDR have been identified by sequence analysis. They have been referred to as LCR-A, -B, -C, and–D.15 They are comprised of smaller modular units which are present in varying arrangements within the LCRs. These chromosome 22-specific LCRs have been reported at or near the end-points of the typical 3 Mb DGS/VCFS/CAFS deletion on 22q11.212,14–19 and at the end-points of the CES duplication.20 This has led to the hypothesis that recombination between copies of the chromosome 22-specific LCRs mediate the deletions and duplications associated with DGS/VCFS/CAFS and CES.15,20–23 The breakpoint of the only recurrent, non-Robertsonian, constitutional translocation involves 22q11 and localizes to LCR-B, one of these chromosome 22-specific LCRs.24–26 The rearrangement, the t(11;22)(q23;q11), has been seen in numerous unrelated families.25,26 Thus, the breakpoints of multiple chromosomal abnormalities involving chromosome 22 appear to localize within the chromosome 22-specific duplications or LCRs. However, although a total of eight LCRs have been identified on 22q11, only the four duplications present within the 3Mb TDR appear to act as recurrent sites for constitutional chromosomal rearrangements.15,23,27 With the exception of the acquired t(9;22) of CML and ALL, which occurs within the BCR gene in another of the 22q11 LCRs, the remaining sites are less frequently involved in rearrangement.
It is clear that the chromosome 22-specific LCRs play a role in mediating these clinically relevant constitutional rearrangements of 22q11. In order to better understand the mechanisms involved in rearrangements associated with 22q11, extensive sequence analysis of the LCRs has been performed. The complicated structure of the LCRs has been refined based on the complete, updated sequence data available for chromosome 22. Using these data, models for the mechanisms involved in the rearrangements of 22q11 are proposed. Furthermore, duplication events in nonhuman primate genomes (chimpanzee, gorilla, rhesus and owl monkey) are demonstrated, suggesting the origin of the LCRs at least 40 million years ago.
COMPLEX ORGANIZATION OF THE 22q11 LCRs
The entire 3 Mb TDR between markers D22S427 and D22S801 has been sequenced at the University of Oklahoma, Advanced Center for Genome Technology (http://www.genome.ou.edu). Sequences of clones within the contig were obtained from the htgs data library of GenBank (http://www.ncbi.nlm.nih.gov). All sequences were masked for repeated DNA elements using the RepeatMasker Web server (http://ftp.genome.washington.edu/cgi-bin/RepeatMasker; Smit and Green, unpublished data). Masked sequences were analyzed further by BLAST searches against the GenBank database28 to identify regions that were duplicated. Sequences from different copies of the duplicated blocks were aligned and compared to each other using ClustalW.29
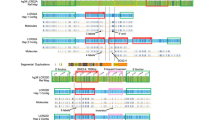
Sequence analysis of the contig spanning LCR -A, -B, -C, and -D has confirmed previous data and demonstrated a somewhat more complex organization of duplicated modules than previously reported (15, Fig. 1). The global differences between LCRs with respect to overall size, content, and organization of duplicated modules within each of them is as described previously.15 Each of the LCRs contains one or more duplicated modules which, in turn, contain previously described duplicated markers, including BCRL, HMPLPL (POM121L), GGTL, NF1L, E2F6L, VNTRL, and ATRRs.15 Although the LCRs differ in content and organization of shared modules, those modules that are common between them share 97–98% sequence identity with one another.15

LCR organization. The spatial arrangement of duplicated modules within LCRs A, B, C, and D is shown. The duplicated modules are shown as colored boxes, and the markers within them are shown above in the same color as the boxes. The orientation of each LCR is centromere to telomere. The sizes of the boxes are proportional to the estimated size of the respective module. Arrows below the duplicated modules indicate their orientation with respect to other copies within the same LCR as well as in other LCRs. The BAC/PAC/cosmid contig spanning each one of the LCRs is shown below each block. Unique markers flanking the LCRs are shown in black. Vertical dashed lines in B mark the boundaries of the gap in LCR-B.
Previous analysis had suggested that the module containing markers D22S131, VNTRL, 562f10Sp6, and NF1L (Fig. 1, green boxes) were all 45 kb in size.15 Closer examination of the sequence has determined that the size of the copies of this module present in the different LCRs can vary. Therefore, in LCR-A, the centromeric and telomeric copies of this module are 35 kb while the one in the middle is 45 kb. Both copies of this module in LCR-D are 45 kb. The copies of this module in LCR-B cannot be correctly estimated due to a remaining gap in the sequence (Fig. 1B). The PI4KL module duplicated in LCR-D is now confirmed to be 25 kb and is in the same orientation as the PI4K gene in LCR-C. LCR-D was previously estimated to be 250 kb in size, but the additional new sequence data suggest that it is larger. Sequence analysis of clones BAC 445f23 and PAC 393h21 has identified additional duplicated modules in LCR-D. Two copies of a 70-kb module (brown boxes in Fig. 1D) have been identified in a head-to-head orientation flanking the PI4KL-containing module. Therefore, the current estimate of LCR-D is 400 kb. LCR-B was previously estimated to be 135 kb based on the existing data.15 Preliminary data from clone cHK89 (Fig. 1B) had suggested that it closed the gap between clones BAC 444p24 and BAC 562f10. Further analysis indicates that, although cHK89 extends the sequence from BAC 444p24, it does not close the gap. Restriction analysis of genomic DNA from normal individuals had indicated the Not I fragment that spans the gap is 145 kb in size.15 Based on these data, the gap in the sequence is estimated to be 90 kb26 making the estimated size of LCR-B at least 225 kb.
UNSTABLE AND UNCLONABLE REGIONS WITHIN LCR-B
The breakpoint of the constitutional t(11;22) on 22q11 localizes to LCR-B within the gap between cHK89 and BAC 562f10.25,26 The breakpoint localizes within an AT-rich repeat (ATRR) that is part of the module containing markers D22S131, VNTRL, 562f10Sp6 and NF1L.26,30 Sequence of the junction fragments obtained from an individual with the constitutional 11;22 translocation suggested that the breakpoint is not within cHK89.26 Furthermore, the sequence at the chromosome 22 breakpoint shares high homology (99%) with duplicated NF1L sequence present in multiple LCRs. This NF1L sequence has similarity to sequence previously implicated in the t(17;22) breakpoint of a patient with neurofibromatosis 1 (NF1).31 Therefore, this suggests that an additional copy of the module containing marker NF1L is present within the gap. Based on additional sequence analysis, this module is predicted to be in a head-to-head orientation with respect to the one in cHK89 and a tail-to-tail orientation with the one in BAC 562f10 (Fig. 1B). This arrangement of highly homologous duplicated modules is likely to lead to an unstable chromatin configuration.
Our data are further supported by the sequence of a cosmid clone, cos4, in the GenBank database that appears to contain a 40-kb junction fragment from a t(11;22) translocation (Accession no. AC074203). The breakpoints in this junction fragment are also in ATRRs and are very similar to those observed in 42 unrelated 11;22 translocation carriers.26,32 The chromosome 22 region of the junction fragment within cos4 is not identical to cHK89 or any of the other known clones on chromosome 22. This suggests that the t(11;22) breakpoint sequence in cos4 lies within the gap in LCR-B. Confirmation of our hypothesis will depend on the isolation of a gap-spanning clone, which has so far proved intractable due to unclonable and unstable sequences in the region.26 This lends further support to the hypothesis of genomic instability in 22q11.
DUPLICATIONS IN NONHUMAN PRIMATES
Previous studies using fluorescence in situ hybridization (FISH) analysis had indicated the presence of 22q11 duplications in humans and on the orthologous chromosomes in pygmy chimpanzee, gorilla, and one Old World monkey, the rhesus monkey.15 This suggested that the human chromosome 22 duplication events predated the divergence of the great apes from the Old World monkeys, which is estimated to have been at least 20–25 million years ago.33 To confirm the FISH data and to test whether the duplications were present in evolutionarily older primates, we employed a polymerase chain reaction (PCR) -based strategy.
A 432-bp fragment designated TSp1, which localizes within the duplicated module containing markers BCRL, HMPLPL, and E2F6L (Fig. 1, blue boxes), was selected for the PCR-based analysis. BLAST database searches with the sequence of TSp1 confirmed that it is present in seven of the eight LCRs on 22q11. Based on human sequence, a primer pair TSp1-F, 5′-ACCTTGGCCTGATTGAGCACT-3′ and TSp1-R, 5′-TCAACAGCCTGTGTGGTGGCA–3′ was designed. These TSp1 primers were then used to PCR amplify the region from genomic DNA of various primates. PCR products were either sequenced directly or subcloned before sequencing individual clones.
The expected product was obtained from human, pygmy chimpanzee, gorilla, rhesus monkey, and owl monkey but not from galago (Fig. 2A). When individual subclones of the TSp1 PCR product generated from the different nonhuman primates were sequenced multiple sequence variants were detected in each primate tested. The number of sequence variants detected in each primate correlated to the evolutionary age of the species. In each case, the number of variants was greater than could be accounted for by allelic variation at a single-copy locus. Therefore, the owl monkey had at least 6 sequence variants while the chimpanzee had greater than 10 sequence variants. The owl monkey is a New World monkey that diverged at least 40 million years ago (mya), and galago is a prosimian that diverged at least 55 mya (Fig. 2B).33,34 This suggests that the duplication events may predate the divergence of New World monkeys (40 mya). It is possible that the duplications may exist in the galago but that the primers designed from human sequence failed to amplify a product due to greater sequence divergence. It is clear that the 22q11 duplications are primate-specific as there is no evidence of their presence in the rodent genome.15 Thus, we have demonstrated the presence of 22q11 duplications in great apes, Old World monkeys, and New World monkeys, suggesting an origin for the duplications at least 40 mya.

PCR analysis of primates with duplicated marker TSp1. A: Gel electrophoresis results of PCR are shown. PCR conditions were as described15 except the annealing temperature was lowered to 50°C for cross-species amplification. Each lane is labeled to indicate the template DNA tested by PCR. K = KG1, a somatic cell hybrid that contains a single human chromosome 22 in a hamster x human hybrid, Hu = human, Ch = pygmy chimpanzee, Go = gorilla, Rh = rhesus monkey, Om = owl monkey, Ga = galago, and W = negative control with no template DNA. M = 1 kb DNA size marker. B: An evolutionary tree that shows the evolution and divergence of the different primate species used in the analysis in A. The approximate time of divergence of each species is indicated below each fork. mya = million years ago. The pygmy chimpanzee, gorilla, and rhesus monkey fibroblast cell lines were obtained from Coriell Mutant Cell repositories (Camden, NJ). The owl monkey and galago cell lines were kind gifts from Dr. Prescott Deininger.
MODELS FOR CONSTITUTIONAL REARRANGEMENTS OF 22q11.2
A number of models have been previously proposed to explain duplicated sequence-mediated chromosomal rearrangements associated with chromosome 22.15,23 The extensive sequence analysis of the LCRs described as the basis of this update has demonstrated high homology between shared modules within the different LCRs as well as some unusual sequence motifs and configurations. These data have led us to propose several models to explain the various rearrangements of 22q11 (Figs. 3 and 4). There are two possible models for the formation of deletions. The first model would involve an interchromosomal misalignment during meiosis I between the two homologs of chromosome 22. This misalignment might be mediated by the modular units within separated LCRs that are in direct orientation with respect to each other. Subsequent crossing-over would lead to reciprocal deletion and duplication events (Fig. 3A). Although deletions are seen frequently, the reciprocal duplication event is rarely observed.23 This is presumed to be the result of a mild and/or nonspecific phenotype.

Models for duplications and deletions of 22q11. Chromosome 22 is shown as a line. Black and red are used to distinguish the two homologs. Filled circles are used to indicate centromeres. LCRs are shown as blue or green boxes. A: Interchromosomal recombination between the two homologs of chromosome 22 leads to a deletion and duplication. B: Intrachromosomal recombination between LCRs on 22q11 leads to a deletion. C: Interchromosomal recombination between the two homologs of chromosome 22 leads to the formation of a bisatellited CES chromosome. D: Paracentric inversion within one homolog of chromosome 22 followed by recombination within an inversion loop leads to the formation of a CES chromosome.

Model for translocations in 22q11. A: Chromosome 22 is shown as a red line with a filled circle to designate the centromere. LCR-B where most of the 22q11 translocation breakpoints localize is shown as a green box. LCRs A, C and D are shown as gray boxes. B: A magnified view of the region containing LCR-B is shown. The centromeric and telomeric ends are indicated. The palindromic sequences in LCR-B (green lines) are predicted to lead to the formation of a hairpin/cruciform structure on chromosome 22. Mismatched regions within the cruciforms may be prone to nicking by nucleases. C: Chromosome 22 with double-stranded breaks within LCR-B could recombine with another chromosome (chromosome “N”) that has similar double-stranded breaks. This would lead to a translocation between chromosome 22 and chromosome “N” leading to the formation of der22 and a der (“N”).
The second model to explain the formation of the deletions would involve intrachromosomal recombination between the duplicated modules during mitosis or meiosis. In this model, the duplicated modules in inverse orientation with respect to one another might form a “stem-loop” intermediate. Recombination between the duplicated modules forming the “stem” would then lead to the deletion of intervening DNA present within the “loop” (Fig. 3B). By haplotype analysis, both inter- and intrachromosomal recombination events have been reported for the standard 3 Mb deletion as well as rearrangements of other chromosomes.23,35,36 Furthermore, there is evidence for mosaic deletions of 22q11 suggesting that mitotic instability does occur37–39
In CES, a bisatellited chromosome resulting from an inverted duplication of proximal 22q11 is present as a supernumerary chromosome.20 There are two distinct duplications in CES patients, and their breakpoints appear to localize to LCRs A and D, which are the proximal and distal deletion end-points of the 3 Mb DGS/VCFS/CAFS common deletion.20 There are two possible models for the formation of the CES marker chromosome. The first model involves interchromosomal misalignment between the two homologs of chromosome 22 by virtue of duplicated modules within the LCRs that are in opposite orientation with respect to one another. Recombination between these inverted sequences would lead to the formation of a bisatellited chromosome seen in CES (Fig. 3C). This model could explain all three types of CES chromosomes that have been identified.20 Therefore, if the misalignment and recombination occurs between the proximal LCRs on both homologs (as shown in Fig. 3C), it gives rise to a type I CES chromosome. Alternatively, the symmetric type II CES chromosome would result from a misalignment and recombination between the two distal LCRs (green boxes in Fig. 3C). Finally, misalignment and recombination between any one proximal (blue box) and any one distal (green box) LCR would result in the formation of an asymmetric type II CES chromosome.
In the second model, intrachromosomal recombination facilitated by the duplicated modules during mitosis or meiosis could first lead to a paracentric inversion (Fig. 3D) in one of the chromosome 22 homologs. Paracentric inversions mediated by duplicated sequences similar to the one proposed here have been described in the region on Xq28 involved in Emery-Dreifuss muscular dystrophy.40 Subsequently, a single crossover event between paired homologs within the inversion loop would lead to the formation of the duplication/deficiency CES marker chromosome (Fig. 3D). This model might explain the formation of some of the asymmetric CES chromosomes.20 A detailed analysis of the chromosomes 22 in parents of CES patients will help to determine which of these mechanisms predominate.
The chromosome 22 breakpoint of the recurrent, constitutional t(11;22) has been localized to LCR-B.26 Multiple other translocation breakpoints also cluster within LCR-B. These include a number of balanced and unbalanced translocations7,31,41 and a balanced t(1;22).42 This further suggests that LCR-B contains unstable sequences that predispose this region to be involved in translocations. The recent identification of palindromes and ATRRs at the breakpoint of the recurrent, constitutional t(11;22) strongly supports this hypothesis.26,30 Analysis of the available sequence from LCR-B suggests the presence of large palindromes flanking the t(11;22) breakpoint region (Fig. 1B). We propose that these palindromic sequences in LCR-B lead to the formation of hairpins or cruciforms. Unpaired regions within the cruciforms are susceptible to nicking by nucleases. Recombination between the nicked chromosome 22 and any other chromosome with similar nicks would lead to a translocation between the two chromosomes (Fig. 4). Although, the recurrent, constitutional t(11;22) appears to be mediated by such a mechanism, there may be additional factors that facilitate this particular recombination event.26,30
CONSEQUENCES OF CHROMOSOME-SPECIFIC DUPLICATIONS IN THE HUMAN GENOME
Chromosome-specific sequence duplications have now been implicated in a number of disorders that are associated with recurrent chromosomal rearrangements such as deletions, duplications, and inversions.43,44 Recombination between duplicated sequences give rise to many genetic disorders, including Charcot-Marie-Tooth disease type 1A (CMT1A) on 17p11.2,45,46 Prader-Willi/Angelman syndromes on 15q11-q13,47,48 Williams-Beuren syndrome on 7q11.23,49,50 and Smith-Magenis syndrome on 17p11.2.51,52 Our own efforts have directly implicated chromosome 22-specific duplications or LCRs in constitutional rearrangements associated with 22q11, including DGS/VCFS/CAFS, CES, and the constitutional t(11;22).15,20,26 As the accumulating human genome sequence is more carefully scrutinized many more chromosome-specific duplications will probably be identified. Thus, in the future, it is likely that many more instances of chromosome-specific duplications will be shown to be responsible for a variety of genomic disorders. Additional analysis of the duplicated sequences should allow us to trace the evolutionary origin and mechanisms involved in their mobility and expansion in the genome. This will assist in clarification of the role of these sequences as mediators of chromosomal instability and rearrangement. These regions of genomic instability are likely to be major contributors to the burden of cytogenetic abnormalities seen in a clinical setting.
References
Morton NE . Parameters of the human genome. Proc Natl Acad Sci USA 1991; 88: 7474–7476.
Kaplan JC, Aurias A, Julier C, Prieur M . Human chromosome 22. J Med Genet 1987; 24: 65–78.
McDermid HE, Duncan AMV, Brasch KR, Holden JJA, Magenis E, Sheehy R, Burn J, Kardon N, Noel B, Schinzel A . Characterization of the supernumerary chromosome in cat eye syndrome. Science 1986; 232: 646–648.
Fraccaro M, Lindsten J, Ford CE, Iselius L . The 11q;22q translocation: a European collaborative analysis of 43 cases. Hum Genet 1980; 56: 2l–51.
Zackai EH, Emanuel BS . Site-specific reciprocal translocation, t(11;22)(q23;q11) in several unrelated families with 3:1 meiotic disjunction. Am J Med Genet 1980; 7: 507–521.
Iselius L, Lindsten J, Aurias A, Fraccaro M, Bastard C, Bottelli AM, Bui TH, Caufin D, Dalpra L, Delendi N . The 11q;22q translocation: a collaborative study of 20 new cases and analysis of 110 families. Hum Genet 1983; 64: 343–355.
De La Chapelle A, Herva R, Koivisto M, Aula P . A deletion in chromosome 22 can cause DiGeorge syndrome. Hum Genet 1981; 57: 253–256.
Kelley RI, Zackai EH, Emanuel BS, Kistenmacher M, Greenberg F, Punnett HH . The association of the DiGeorge anomalad with partial monosomy of chromosome 22. J Pediatr 1981; 101: 197–200.
Driscoll DA, Budarf ML, Emanuel BS . A genetic etiology for DiGeorge syndrome: consistent deletions and microdeletions of 22q11. Am J Hum Genet 1992; 50: 924–933.
Driscoll DA, Spinner NB, Budarf ML, McDonald-McGinn DM, Zackai EH, Goldberg RB, Shprintzen RJ, Saal HM, Zonana J, Jones MC . Deletions and microdeletions of 22q11.2 in velo-cardio-facial syndrome. Am J Med Genet 1992; 44: 261–268.
Carey AH, Kelly D, Halford S, Wadey R, Wilson D, Goodship J, Burn J, Paul T, Sharkey A, Dumanski J, Nordenskjold M, Williams R, Scambler PJ . Molecular genetic study of the frequency of monosomy 22q11 in DiGeorge syndrome. Am J Hum Genet 1992; 51: 964–970.
Driscoll DA, Salvin J, Sellinger B, Budarf ML, McDonald-McGinn DM, Zackai EH, Emanuel BS . Prevalence of 22q11 microdeletions in DiGeorge, and velocardiofacial syndromes: implications for genetic counselling and prenatal diagnosis. J Med Genet 1993; 30: 813–817.
Burn J, Takao A, Wilson D, Cross I, Momma K, Wadey R, Scambler P, Goodship J . Conotruncal anomaly face syndrome is associated with a deletion within chromosome 22. J Med Genet 1993; 30: 822–824.
Kelley D, Goldberg R, Wilson D, Lindsay E, Carey A, Goodship J, Burn J, Cross I, Shprintzen RJ, Scambler PJ . Confirmation that the velo-cardiofacial syndrome is associated with haplo-insufficiency of genes at chromosome 22. Am J Med Genet 1993; 45: 308–312.
Shaikh TH, Kurahashi H, Saitta SC, O'Hare AM, Hu P, Roe BA, Driscoll DA, McDonald-McGinn DM, Zackai EH, Budarf ML, Emanuel BS . Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: genomic organization and deletion endpoint analysis. Hum Mol Genet 2000; 9: 489–501.
Halford S, Lindsay E, Natudu M, Carey AH, Baldini A, Scambler PJ . Low-copy-number repeat sequences flank the DiGeorge/velo-cardio-facial syndrome loci at 22q11. Hum Mol Genet 1993; 2: 191–196.
Lindsay EA, Greenberg F, Shaffer LG, Scamber PJ, Baldini A . Submicroscopic deletions at 22q11.2: variability of the clinical picture and delineation of a common deleted region. Am J Med Genet 1995; 56: 191–197.
Morrow B, Goldberg R, Carlson C, Dasgupta R, Sirotkin H, Collins J, Dunham I, O'Donnell H, Scambler P, Shprintzen R . Molecular definition of the 22q11 deletions in velo-cardio-facial syndrome. Am J Hum Genet 1995; 56: 1391–1403.
Collins JE, Mungall AJ, Badcock KL, Fay JM, Dunham I . The organization of gamma-glutamyltransferase genes and other low copy repeats in human chromosome 22q11. Genome Res 1997; 7: 522–531.
McTaggart KE, Budarf ML, Driscoll DA, Emanuel BS, Ferreira P, McDermid HE . Cat eye syndrome chromosome breakpoint clustering: identification of two inter-vals also associated with 22q11 deletion syndrome breakpoints. Cytogenet Cell Genet 1998; 81: 222–228.
Emanuel BS, Goldmuntz E, Budarf ML, Shaikh T, McGrath J, McDonald-McGinn D, Zackai EH . et al. Blocks of duplicated sequence define the endpoints of DGS/VCFS 22q11.2 deletions. In: Clark E, Nakazawa M, Takao A, editors. Etiology and morphogenesis of congenital heart disease. Armonk, NY: Futura Publishing Company, Inc., 1999.
Edelmann L, Pandita RK, Morrow BE . Low-copy repeats mediate the common 3-Mb deletion in patients with velo-cardio-facial syndrome. Am J Hum Genet 1999; 64: 1076–1086.
Edelmann L, Pandita RK, Spiteri E, Funke B, Goldberg R, Palaiswamy N, Chaganti RSK, Magenis E, Shprintzen RJ, Morrow BE . A common molecular basis for rearrangement disorders on chromosome 22q11. Hum Mol Genet 1999; 8: 1157–1167.
Funke B, Edelmann L, McCain N, Pandita RK, Ferreira J, Merscher S, Zohouri M, Cannizzaro L, Shanske A, Morrow BE . Der(22) syndrome, and Velo-Cardio-Facial syndrome/DiGeorge syndrome share a 1.5-Mb region of overlap on chromosome 22q11. Am J Hum Genet 1999; 64: 747–758.
Shaikh TH, Budarf ML, Celle L, Zackai EH, Emanuel BS . Clustered 11q23, and 22q11 breakpoints, and 3. 1 meiotic malsegregation in multiple unrelated t(11;22) families. Am J Hum Genet 1999; 65: 1595–1607.
Kurahashi H, Shaikh TH, Hu P, Roe B, Emanuel BS, Budarf ML . Regions of genomic instability on 22q11, and 11q23 as the etiology for the recurrent constitutional t(11;22). Hum Mol Genet 2000; 9: 1665–1670.
Dunham I, Shimizu N, Roe BA, Chissoe S, Hunt AR, Collins JE, Bruskiewich R, Beare DM, Clamp M, Smink LJ, Ainscough R, Almeida JP, Babbage A, Bagguley C, Bailey J, Barlow K, Bates KN, Beasley O, Bird CP, Blakey S, Bridgeman AM, Buck D, Burgess J, Burrill WD, O'Brien KP . The DNA sequence of human chromosome 22. Nature 1999; 402: 489–495.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ . Basic local alignment search tool. J Mol Biol 1990; 215: 403–410.
Thompson JD, Higgins DG, Gibson TJ . CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties and weight matrix choice. Nucleic Acids Res 1994; 22: 4673–4680.
Edelmann L, Spiteri E, McCain N, Goldberg R, Pandita RK, Duong S, Fox J, Blumenthal D, Lalani SR, Shaffer LG, Morrow BE . A common breakpoint on 11q23 in carriers of the constitutional t(11;22) translocation. Am J Hum Genet 1999; 65: 1608–1616.
Kehrer-Sawatzki H, Haussler J, Krone W, Bode H, Jenne DE, Mehnert KU, Tummers U, Assum G . The second case of a t(17;22) in a family with neurofibromatosis type 1: sequence analysis of the breakpoint regions. Hum Genet 1997; 99: 237–247.
Kurahashi H, Shaikh TH, Zackai EH, Celle L, Driscoll DA, Budarf ML, Emanuel BS . Tightly Clustered 11q23, and 22q11 Breakpoints Permit PCR-Based Detection of the Recurrent Constitutional t(11;22). Am J Hum Genet 2000; 67: 763–768.
Kumar S, Hedges BS . A molecular timescale for vertebrate evolution. Nature 1998; 392: 917–920.
Shen MR, Batzer MA, Deininger PL . Evolution of the Master Alu Gene(s). J Mol Evol 1991; 33: 311–320.
Baumer A, Dutly F, Balmer D, Riegel M, Tukel T, Krajewska-Walasek M, Schinzel AA . High level of unequal meiotic crossovers at the origin of the 22q11.2 and 7q11.23 deletions. Hum Mol Genet 1998; 7: 887–894.
Saitta SC, Driscoll DA, Rappaport EF, McDonald-McGinn D, Zackai EH, Emanuel BS . The 22q11.2 deletion: parental origin and meiotic mechanism. Am J Hum Genet 2000; 67: 163.
Consevage MW, Seip JR, Belchis DA, Davis AT, Baylen BG, Rogan PK . Association of a mosaic chromosomal 22q11 deletion with hypoplastic left heart syndrome. Am J Cardiol 1996; 77: 1023–1025.
Kasprzak L, Der Kaloustian VM, Elliott AM, Shevell M, Lejtenyi C, Eydoux P . Deletion of 22q11 in two brothers with different phenotype. Am J Med Genet 1998; 75: 288–291.
Anguiano A, Hudgins L, Garscadden A, Mak L, Tayag A, Emanuel BS . First report of a case with manifestations of DiGeorge/Velocardiofacial syndrome (VCFS) and microdeletion 22q11.2 mosaicism. Am J Hum Genet. 2000 ( in press).
Small K, Iber J, Warren ST . Emerin deletion reveals a common X-chromosome inversion mediated by inverted repeats. Nat Genet 1997; 16: 96–99.
Li M, Budarf ML, Chien P, Barnoski BL, Emanuel BS, Driscoll DA . Clustering of DiGeorge/velocardiofacial-associated translocations suggestive of a translocation “hot spot.”. Am J Hum Genet 1995; 57: A119.
Rhodes CH, Call KM, Budarf ML, Barnoski BL, Bell CJ, Emanuel BS, Bigner SH . Molecular studies of an ependymoma-associated constitutional t(1;22)(p22;q11.2). Cytogenet Cell Genet 1997; 78: 247–252.
Lupski JR . Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet 1998; 14: 417–422.
Mazzarella R, Schlessinger D . Pathological consequences of sequence duplications in the human genome. Genome Res 1998; 8: 1007–1021.
Chance PF, Abbas N, Lensch MW, Pentao L, Rao BB, Patel PI, Lupski JR . Two autosomal dominant neuropathies result from reciprocal DNA duplication/deletion of a region on chromosome 17. Hum Mol Genet 1994; 3: 223–228.
Lupski JR . Charcot-Marie-Tooth disease: lessons in genetic mechanisms. Mol Med 1998; 4: 3.
Christian SL, Fantes JA, Mewborn SK, Huang B, Ledbetter DH . Large genomic dulicons map to sites of instability in Prader-Willi/Angelman syndrome chromosome region (15q11-q13). Hum Mol Genet 1990; 8: 1025–1037.
Amos-Landgraf JM, Ji Y, Gottlieb W, Depinet T, Wandstrat AE, Cassidy SB, Driscoll DJ, Rogan PK, Schwartz S, Nicholls RD . Chromosome breakage in the Prader-Willi and Angelman syndromes involves recombination between large, transcribed repeats at proximal and distal breakpoints. Am J Hum Genet 1999; 65: 370–386.
Perez -Jurado LA, Wang YK, Peoples R, Coloma A, Cruces J, Francke U . A duplicated gene in the breakpoint regions of the 7q11.23 Williams-Beuren syndrome deletion encodes the initiator binding protein TFII-I and BAP-135, a phosphorylation target of BTK. Hum Mol Genet 1998; 7: 325–334.
Peoples R, Franke Y, Wang YK, Perez-Jurado L, Paperna T, Cisco M, Francke U . A physical map, including a BAC/PAC clone contig, of the Williams-Beuren syndrome–deletion region at 7q11.23. Am J Hum Genet 2000; 66: 47–68.
Chen KS, Manian P, Koeuth T, Potocki L, Zhao Q, Chinault AC, Lee CC . Homologous recombination of a flanking repeat gene cluster is a mechanism for a common contiguous gene deletion syndrome. Nat Genet 1997; 17: 154–163.
Potocki L, Chen KS, Park SS, Osterholm DE, Withers MA, Kimonis V, Summers AM, Meschino WS, Anyane-Yeboa K, Kashork CD, Shaffer LG, Lupski JR . Molecular mechanism for duplication 17p11.2- the homologous recombination reciprocal of the Smith-Magenis microdeletion. Nat Genet 2000; 24: 84–87.
Acknowledgements
This work was supported by grants CA39926 and DC02027 (B.S.E.), HD26979 (B.S.E.), and funds from the Charles E. H. Upham endowed chair from The Children’s Hospital of Philadelphia and the University of Pennsylvania (B.S.E.).
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Shaikh, T., Kurahashi, H. & Emanuel, B. Evolutionarily conserved low copy repeats (LCRs) in 22q11 mediate deletions, duplications, translocations, and genomic instability: An update and literature review. Genet Med 3, 6–13 (2001). https://doi.org/10.1097/00125817-200101000-00003
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/00125817-200101000-00003
Keywords
This article is cited by
-
Diagnosis of prenatal 22q11.2 duplication syndrome: a two-case study
Journal of Genetics (2022)
-
Two novel mouse models mimicking minor deletions in 22q11.2 deletion syndrome revealed the contribution of each deleted region to psychiatric disorders
Molecular Brain (2021)
-
In the line-up: deleted genes associated with DiGeorge/22q11.2 deletion syndrome: are they all suspects?
Journal of Neurodevelopmental Disorders (2019)
-
Recurrent pyoderma gangrenosum precipitated by breast cancer: a case report and review of the literature
Journal of Medical Case Reports (2014)
-
Mapping the deletion endpoints in individuals with 22q11.2 Deletion Syndrome by droplet digital PCR
BMC Medical Genetics (2014)
