
Abstract
The CC chemokine receptor 5 (CCR5) has been shown to be important in the recruitment of T-helper cells to the synovium, where they accumulate, drive the inflammatory process and the consequent synovitis and joint destruction. A 32 base-pair insertion/deletion variant (CCR5Δ32) within the gene leads to a frame shift and a nonfunctional receptor. CCR5Δ32 has been investigated for its association with juvenile idiopathic arthritis (JIA), with conflicting results. The aim of this study was to investigate whether CCR5Δ32 is associated with JIA in an UK population. CCR5Δ32 was genotyped in JIA cases (n=1054) and healthy controls (n=3129) and genotype and allele frequencies were compared. A meta-analysis of our study combined with previously published studies was performed. CCR5Δ32 was significantly associated with protection from developing JIA, in this UK data set (Ptrend=0.006, odds ratio (OR) 0.79 95% confidence interval (95% CI): 0.66–0.94). The meta-analysis of all published case–control association studies confirmed the protective association with JIA (P=0.001 OR 0.82 95% CI: 0.73–0.93). CCR5Δ32 is a functional variant determining the number of receptors on the surface of T cells, and it is hypothesized that the level of CCR5 expression could influence the migration of proinflammatory T cells into the synovium and thus susceptibility to JIA.
Similar content being viewed by others

Introduction
Juvenile idiopathic arthritis (JIA) is the most common chronic rheumatic disease of childhood. It is defined as chronic inflammation of the synovial joints, with unknown etiology, which starts before the age of 16 years and persists for at least 6 weeks. There is a wealth of evidence to suggest that the pathogenesis is largely driven by T cells.1 It has been shown that there is a strong bias in the overall T-helper (Th) 1 cytokine profile of synovial fluid (SF) T cells compared with peripheral blood T cells from patients with either oligoarticular or polyarticular JIA, and it is proposed that this polarization may be caused by selective recruitment by chemokine receptor expression.2 CC chemokine receptor 5 (CCR5) is predominantly found on monocytes and T cells, particularly of the Th1 phenotype.3 Its function is in the recruitment of these Th1 T cells to the synovium, where they accumulate and produce proinflammatory cytokines, such as interleukin-2 (IL2) and interferon-γ (which drives the inflammatory process and the consequent synovitis and joint destruction). CCR5 binds a number of proinflammatory chemokines, including CC chemokines, such as CCL3 (also known as macrophage inflammatory protein 1α), CCL4 (macrophage inflammatory protein β) and CCL5 (also known as regulated on activation, normal T-cell expressed and secreted).4 These chemokines have been found at high levels in the SF of patients with adult rheumatoid arthritis (RA),5 suggesting that CCR5+ T cells selectively accumulate in the SF in response to the presence of these chemokines. Furthermore, it has also been noted that the cell-surface CCR5 density determines the intensity of T-cell migration toward RA synoviocytes and also their stimulation by CCL5.6 Thus, the level of CCR5 expression could favor the inflammatory effect of intra-articular T cells by facilitating their recruitment and increasing their response to chemokines. Therefore, the density of CCR5 on cells may be predictive of disease severity.
The CCR5 gene on chromosome 3p21.3 contains a 32 base-pair insertion/deletion variant (CCR5Δ32), which leads to a frame shift and a nonfunctional receptor. Homozygosity for CCR5Δ32 results in a complete lack of surface expression of CCR5, whereas heterozygotes express low densities of cell-surface CCR5 compared with individuals homozygous for the wild-type CCR5 gene.7 The polymorphism has been extensively studied in RA with conflicting evidence for a protective association of the CCR5Δ32 polymorphism.8, 9, 10, 11, 12 However, a large meta-analysis of five case–control association studies confirmed the protective association between CCR5Δ32 and RA (odds ratio (OR) 0.65 95% confidence interval (95% CI): 0.55–0.77, P<0.0001).13 In JIA, again, the results are conflicting; one study reported that the CCR5Δ32 variant was protectively associated with JIA;14 another described no association;15 and a third found that the CCR5Δ32 variant was associated with JIA susceptibility.16 The aim of this study was to investigate whether CCR5Δ32 is associated with JIA in an UK population and to combine the results with those obtained in previous studies by meta-analysis.
Results
Genotype counts conformed to Hardy–Weinberg expectations (P>0.05) in the control cohort. After quality control, 987 JIA samples and 3121 control samples were available for analysis. We found that the CCR5Δ32 variant was significantly associated with protection from developing JIA (Ptrend=0.006, OR 0.79 95% CI: 0.66–0.94) (Table 1).
Interestingly, the strongest protective effect was found in the rheumatoid factor-positive polyarthritis subtype (OR 0.41 95% CI: 0.18–0.93) (Table 2); however, there was no significant difference in allele frequencies across all the subtypes, suggesting that no subtype-specific associations exist.
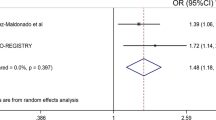
To perform the meta-analysis, allele and genotype data from previous studies in JIA were collated (Table 3). Three previous studies were identified from literature searches: one from the United States,14 the second, a study of Norwegian JIA cases and controls15 and finally a study of Brazilian JIA cases and controls.16 The US study was a family-based association analysis; hence for the meta-analysis, data collected from the parents and unaffected siblings were used as control data. Studies from the Unites States and Norway reported very similar control allele frequencies (10–11%). However, control allele frequencies in the Brazilian study by Scheibel et al. were very different (3.8%). This study was relatively small and reported association of the CCR5Δ32 variant with JIA susceptibility. Inclusion of this study in the meta-analysis resulted in a P-value of 0.04 for the Breslow–Day test, suggesting significant heterogeneity between the studies. In view of the heterogeneity, the meta-analysis was, therefore, restricted to cohorts of Northern European descent and the results from the Brazilian cohort were not included in the calculation of the pooled OR. The pooled OR for the UK, US and Norwegian studies (Breslow–Day P=0.74) was 0.82 95% CI: 0.73–0.93, P=0.001. The combined analysis had a slightly more significant P-value and tighter CIs than that found for our UK JIA study alone (Figure 1).

Meta-analysis of three studies of the CCR5Δ32 variant. Meta-analysis of UK, US and Norwegian JIA cohorts for the CCR5 gene. Forest plot displaying odds ratios and 95% confidence intervals for each of the studies and the weighting for each study according to sample size. The combined OR from the three studies is displayed as a diamond with the peaks denoting the upper and lower limits of the confidence intervals. Values below 1 depict a negative (protective) association. The Breslow-Day test was performed to test for heterogeneity between the two studies, with a significant P-value suggesting heterogeneity between studies. The combined P-value is displayed below the heterogeneity P-value.
Discussion
The results from this investigation and the meta-analysis of previous studies suggest that the CCR5Δ32 variant is associated with protection from developing JIA for individuals of Northern European descent. Our study was the largest to date to investigate this association and had >80% power to detect a protective OR of ⩽0.75, similar to the effect size identified in the Prahalad meta-analysis for RA (OR=0.65).13 The meta-analysis highlights the need for large sample sizes to detect variants with a modest effect.
The US family-based association study of the CCR5Δ32 variant found no significant deviation of transmission of CCR5Δ32 in the combined cohort of probands with JIA. However, stratification by age at onset found that CCR5Δ32 was significantly undertransmitted to probands with early-onset JIA (<6 years). In the UK cohort, there was no evidence for a stronger effect in cases with a young age at onset (Ptrend=0.1, OR 0.83 95% CI: 0.66–1.04); in fact, those with an older age at onset showed a slightly stronger effect (Ptrend=0.03, OR 0.77 95% CI: 0.60–0.97).
The same protective association has been found in RA but, as with JIA, there have been some conflicting results; however, a meta-analysis of five case–control association studies confirmed the protective association between CCR5Δ32 and RA (OR 0.65 95% CI: 0.55–0.77, P<0.0001). This is further evidence for shared susceptibility loci for RA and JIA, and adds to the evidence from other loci, which are associated with both diseases (and often other autoimmune diseases as well), such as PTPN22, IL2/IL21 region, IL2RA, AFF3, STAT4, TRAF1/C5 and TNFAIP3.17, 18, 19 Indeed, the CCR5Δ32 variant has previously been associated with protection from developing both type I diabetes and celiac disease,20 suggesting a pan-autoimmune disease role.
The influence of CCR5Δ32 on the clinical course of RA has also been investigated, but again the evidence is often conflicting, with the most recent study finding that carriers of the deletion were protected from joint erosion, had less extra-articular manifestations and had lower cumulative C-reactive protein levels.21 However, a number of other studies have found no evidence for an association with disease severity or outcome.10, 22, 23 These discrepancies may arise as a result of differences in statistical power and methodologies used but, given the fact that CCR5 expression levels may be associated with severity, it would be interesting to investigate whether CCR5Δ32 has any effect on outcome in JIA. Data from the CAPS (Childhood Arthritis Prospective Study), a prospective inception cohort study of JIA cases, will be invaluable in exploring this hypothesis, but are insufficiently powered at present to undertake these investigations.
The observation that there are six JIA cases that are homozygous for CCR5Δ32 suggests that there may be some redundancy in the chemokine system and other receptors and ligands may have a role in the development of JIA. A previous study that found high expression of two chemokine receptors, CCR5 and CXCR3 on SF on T cells from 20 JIA patients compared with peripheral blood T cells also found some evidence of redundancy.2 In that study, one of the JIA patients was homozygous for CCR5Δ32 but was, perhaps surprisingly, of the more severe extended oligoarthritis subtype. This individual had high levels of CXCR3 on SF T cells and had a high interferon-γ:IL4 ratio in the cytokine stimulation assay. In most cases, SF T cells were double positive for CCR5 and CXCR3, but T cells can express one or the other. The different coexpression also suggests that the levels of these two receptors are regulated by different factors.
It has been proposed that CCR5 is important in the recruitment of Th1 T cells to the joint but also, interestingly, a number of studies have linked CCR5 to the IL2 pathway. For example, IL2 upregulates CCR5 expression in vivo and in vitro24, 25, 26 and rapamycin, a drug that disrupts IL2 receptor signaling, reduces CCR5 surface expression on T cells.27 A recent study found that CCR5Δ32/Δ32 donors of peripheral blood mononuclear cells stimulated by antiCD3/CD28 showed significantly less IL2 production than did noncarriers of the CCR5Δ32 mutation. They showed that CCR5 influenced IL2 transcript levels and also that CCR5 influenced the expression of IL2RA/CD25 on CD4+ T cells both in vitro and in vivo. There was evidence that these effects are mediated through the nuclear factor of activated T cell signaling pathway. CCR5 expression was also shown to promote T-cell proliferation after T-cell activation.28 This is fascinating in that the IL2 gene region19, 29 and IL2RA genes30 are also associated with JIA. Interestingly, in all three cases, the SNP in the gene/region is associated with protection from JIA.
In conclusion, we have presented evidence for a protective association of the CCR5Δ32 variant in an UK JIA case–control cohort, which is strengthened by combining data obtained from two previous studies in populations of Northern European descent by meta-analysis. Further investigation into how this gene contributes to JIA pathogenesis is now required.
Materials and methods
Subjects
DNA was available for 1054 UK Caucasian JIA patients (332 males and 715 females) from three sources: BSPAR (The British Society for Paediatric and Adolescent Rheumatology) National Repository of JIA (n=654); a group of UK Caucasian patients with long-standing JIA (n=201), described previously;31 and a third cohort collected as part of the CAPS, a prospective inception cohort study of JIA cases from five centers across the United Kingdom (n=199).32 JIA cases were classified according to the ILAR criteria33 (Supplementary Table 1). Healthy Caucasian control DNA samples (n=3129) were available from five centers in the United Kingdom as described previously:34 Manchester, Sheffield, Leeds, Aberdeen and Oxford (total control sample size after quality control=3121). All individuals were recruited with ethical approval and provided informed consent (North-west Multi-Centre Research Ethics Committee (MREC 99/8/84) and the University of Manchester Committee on the Ethics of Research on Human Beings).
Genotyping
The CCR5Δ32 variant (rs333) was genotyped using the Sequenom iPlex MassARRAY platform according to the manufacturer's instructions (Sequenom, San Diego, CA, USA; http://www.sequenom.com/). A 90% sample quality control rate and a 90% genotyping success rate were imposed on the analysis.
Statistical analysis
Power calculations were performed using QUANTO to calculate the prior probability of detecting association in the current sample size at the allele frequency and effect sizes were reported previously in other autoimmune diseases for this variant.14 Calculations assumed a log-additive model, an α value of 0.05 and an effect size of 0.75. Genotype and allele frequencies were compared between cases with JIA and controls using the Cochrane–Armitage trend test implemented in PLINK (http://pngu.mgh.harvard.edu/purcell/plink/),35 and allelic ORs and their 95% CIs were calculated. JIA is a phenotypically heterogeneous disease and can be classified into more clinically homogeneous diseases using the ILAR classification criteria.36 However, comparing each of the ILAR subtypes separately against controls would result in a large number of hypothesis tests. Therefore, we also examined whether there was evidence of a difference in allele frequencies between the seven ILAR subtypes. Differences between subtypes were assessed using χ2-tests on the 7 × 2 tables.
Published case–control association studies of CCR5Δ32 with JIA were identified using a PUBMED search. Genotype and allele frequencies were obtained from the published studies. A test for heterogeneity between cohorts was carried out using the Breslow–Day test, and the Cochran–Mantel–Haenszel test was used to perform a meta-analysis of the three cohorts.
References
Murray K, Thompson SD, Glass DN . Pathogenesis of juvenile chronic arthritis: genetic and environmental factors. Arch Dis Child 1997; 77: 530–534.
Wedderburn LR, Robinson N, Patel A, Varsani H, Woo P . Selective recruitment of polarized T cells expressing CCR5 and CXCR3 to the inflamed joints of children with juvenile idiopathic arthritis. Arthritis Rheum 2000; 43: 765–774.
Loetscher P, Uguccioni M, Bordoli L, Baggiolini M, Moser B, Chizzolini C et al. CCR5 is characteristic of Th1 lymphocytes. Nature 1998; 391: 344–345.
Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P . Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science 1995; 270: 1811–1815.
Suzuki N, Nakajima A, Yoshino S, Matsushima K, Yagita H, Okumura K . Selective accumulation of CCR5+ T lymphocytes into inflamed joints of rheumatoid arthritis. Int Immunol 1999; 11: 553–559.
Desmetz C, Lin YL, Mettling C, Portales P, Noel D, Clot J et al. Cell surface CCR5 density determines the intensity of T cell migration towards rheumatoid arthritis synoviocytes. Clin Immunol 2007; 123: 148–154.
Venkatesan S, Petrovic A, Van Ryk DI, Locati M, Weissman D, Murphy PM . Reduced cell surface expression of CCR5 in CCR5Delta 32 heterozygotes is mediated by gene dosage, rather than by receptor sequestration. J Biol Chem 2002; 277: 2287–2301.
Garred P, Madsen HO, Petersen J, Marquart H, Hansen TM, Freiesleben SS et al. CC chemokine receptor 5 polymorphism in rheumatoid arthritis. J Rheumatol 1998; 25: 1462–1465.
Gomez-Reino JJ, Pablos JL, Carreira PE, Santiago B, Serrano L, Vicario JL et al. Association of rheumatoid arthritis with a functional chemokine receptor, CCR5. Arthritis Rheum 1999; 42: 989–992.
Pokorny V, McQueen F, Yeoman S, Merriman M, Merriman A, Harrison A et al. Evidence for negative association of the chemokine receptor CCR5 d32 polymorphism with rheumatoid arthritis. Ann Rheum Dis 2005; 64: 487–490.
Zapico I, Coto E, Rodriguez A, Alvarez C, Torre JC, Alvarez V . CCR5 (chemokine receptor-5) DNA-polymorphism influences the severity of rheumatoid arthritis. Genes Immun 2000; 1: 288–289.
Cooke SP, Forrest G, Venables PJ, Hajeer A . The delta32 deletion of CCR5 receptor in rheumatoid arthritis. Arthritis Rheum 1998; 41: 1135–1136.
Prahalad S . Negative association between the chemokine receptor CCR5-Delta32 polymorphism and rheumatoid arthritis: a meta-analysis. Genes Immun 2006; 7: 264–268.
Prahalad S, Bohnsack JF, Jorde LB, Whiting A, Clifford B, Dunn D et al. Association of two functional polymorphisms in the CCR5 gene with juvenile rheumatoid arthritis. Genes Immun 2006; 7: 468–475.
Lindner E, Nordang GB, Melum E, Flato B, Selvaag AM, Thorsby E et al. Lack of association between the chemokine receptor 5 polymorphism CCR5delta32 in rheumatoid arthritis and juvenile idiopathic arthritis. BMC Med Genet 2007; 8: 33.
Scheibel I, Veit T, Neves AG, Souza L, Prezzi S, Machado S et al. Differential CCR5Delta32 allelic frequencies in juvenile idiopathic arthritis subtypes: evidence for different regulatory roles of CCR5 in rheumatological diseases. Scand J Rheumatol 2008; 37: 13–17.
Hinks A, Worthington J, Thomson W . The association of PTPN22 with rheumatoid arthritis and juvenile idiopathic arthritis. Rheumatology (Oxford) 2006; 45: 365–368.
Hinks A, Eyre S, Ke X, Barton A, Martin P, Flynn E et al. Overlap of disease susceptibility loci for rheumatoid arthritis (RA) and juvenile idiopathic arthritis (JIA). Ann Rheum Dis (e-pub ahead of print 11 August 2009).
Hinks A, Eyre S, Ke X, Barton A, Martin P, Flynn E et al. Association of the AFF3 gene and IL2/IL21 gene region with juvenile idiopathic arthritis. Genes Immun 2010; 11: 194–198.
Smyth DJ, Plagnol V, Walker NM, Cooper JD, Downes K, Yang JH et al. Shared and distinct genetic variants in type 1 diabetes and celiac disease. N Engl J Med 2008; 359: 2767–2777.
Rossol M, Pierer M, Arnold S, Keysser G, Burkhardt H, Baerwald C et al. Negative association of the chemokine receptor CCR5 d32 polymorphism with systemic inflammatory response, extra-articular symptoms and joint erosion in rheumatoid arthritis. Arthritis Res Ther 2009; 11: R91.
Zuniga JA, Villarreal-Garza C, Flores E, Barquera R, Perez-Hernandez N, Montes de Oca JV et al. Biological relevance of the polymorphism in the CCR5 gene in refractory and non-refractory rheumatoid arthritis in Mexicans. Clin Exp Rheumatol 2003; 21: 351–354.
John S, Smith S, Morrison JF, Symmons D, Worthington J, Silman A et al. Genetic variation in CCR5 does not predict clinical outcome in inflammatory arthritis. Arthritis Rheum 2003; 48: 3615–3616.
Yang YF, Tomura M, Iwasaki M, Mukai T, Gao P, Ono S et al. IL-12 as well as IL-2 upregulates CCR5 expression on T cell receptor-triggered human CD4+ and CD8+ T cells. J Clin Immunol 2001; 21: 116–125.
Zou W, Foussat A, Houhou S, Durand-Gasselin I, Dulioust A, Bouchet L et al. Acute upregulation of CCR-5 expression by CD4+ T lymphocytes in HIV-infected patients treated with interleukin-2. ANRS 048 IL-2 Study Group. AIDS 1999; 13: 455–463.
Weissman D, Dybul M, Daucher MB, Davey Jr RT, Walker RE, Kovacs JA . Interleukin-2 up-regulates expression of the human immunodeficiency virus fusion coreceptor CCR5 by CD4+ lymphocytes in vivo. J Infect Dis 2000; 181: 933–938.
Heredia A, Amoroso A, Davis C, Le N, Reardon E, Dominique JK et al. Rapamycin causes down-regulation of CCR5 and accumulation of anti-HIV beta-chemokines: an approach to suppress R5 strains of HIV-1. Proc Natl Acad Sci USA 2003; 100: 10411–10416.
Camargo JF, Quinones MP, Mummidi S, Srinivas S, Gaitan AA, Begum K et al. CCR5 expression levels influence NFAT translocation, IL-2 production, and subsequent signaling events during T lymphocyte activation. J Immunol 2009; 182: 171–182.
Albers HM, Kurreeman FA, Stoeken-Rijsbergen G, Brinkman DM, Kamphuis SS, Van Rossum MA et al. Association of the autoimmunity locus 4q27 with juvenile idiopathic arthritis. Arthritis Rheum 2009; 60: 901–904.
Hinks A, Ke X, Barton A, Eyre S, Bowes J, Worthington J et al. Association of the IL2RA/CD25 gene with juvenile idiopathic arthritis. Arthritis Rheum 2009; 60: 251–257.
Packham JC, Hall MA . Long-term follow-up of 246 adults with juvenile idiopathic arthritis: functional outcome. Rheumatology (Oxford) 2002; 41: 1428–1435.
Adib N, Hyrich K, Thornton J, Lunt M, Davidson J, Gardner-Medwin J et al. Association between duration of symptoms and severity of disease at first presentation to paediatric rheumatology: results from the Childhood Arthritis Prospective Study. Rheumatology (Oxford) 2008; 47: 991–995.
Petty RE, Southwood TR, Baum J, Bhettay E, Glass DN, Manners P et al. Revision of the proposed classification criteria for juvenile idiopathic arthritis: Durban, 1997. J Rheumatol 1998; 25: 1991–1994.
Thomson W, Barton A, Ke X, Eyre S, Hinks A, Bowes J et al. Rheumatoid arthritis association at 6q23. Nat Genet 2007; 39: 1431–1433.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Petty RE, Southwood TR, Manners P, Baum J, Glass DN, Goldenberg J et al. International League of Associations for Rheumatology Classification of Juvenile Idiopathic Arthritis: second revision, Edmonton, 2001. J Rheumatol 2004; 31: 390–392.
Acknowledgements
This work was supported by the Arthritis Research UK: grant reference no: 17552. We thank David Strachan for facilitating access to the 1958 birth cohort. We acknowledge the use of genotype data from the British 1958 Birth Cohort DNA collection, funded by the Medical Research Council grant G0000934 and the Wellcome Trust grant 068545/Z/02. We acknowledge support from the NIHR Manchester Biomedical Research Council.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Genes and Immunity website
Supplementary information
Appendix
Appendix
Childhood arthritis prospective study (CAPS):
Eileen Baildam, Lynsey Brown, Joanne Buckley, Alice Chieng, Joyce Davidson, Michael Eltringham, Helen Foster, Mark Friswell, Janet Gardner-Medwin, Paul Gilbert, Kimme Hyrich, Julie Jones, Sham Lal, Mark Lay, Carol Lydon, Alexandra Meijer, Vicki Price, Jane Sim, Maureen Todd, Peter Ward, Lucy Wedderburn.
UKRAG Consortium:
University of Manchester1: Stephen Eyre, Anne Hinks, Laura J Gibbons, John Bowes, Edward Flynn, Paul Martin, Xiayi Ke, Rachelle Donn, Wendy Thomson, Anne Barton, Jane Worthington.
University of Leeds2: Stephen Martin, James I Robinson, Ann W Morgan, Paul Emery
University of Sheffield3: Anthony G Wilson
University of London4: Sophia Steer
University of Aberdeen5: Lynne Hocking, David M Reid
University of Oxford6: Pille Harrison, Paul Wordsworth
1arc-Epidemiology Unit, Stopford Building, The University of Manchester, Manchester, UK.
2Leeds Musculoskeletal Biomedical Research Unit, Leeds Institute of Molecular Medicine, University of Leeds, Leeds, LS9 7TF.
3School of Medicine and Biomedical Sciences, The University of Sheffield, Sheffield S10 2JF.
4Clinical and Academic Rheumatology, Kings College Hospital NHS Foundation Trust, Denmark Hill, London SE5 9RS.
5Bone Research Group, Department of Medicine and Therapeutics, University of Aberdeen, UK, AB25 2ZD
6University of Oxford Institute of Musculoskeletal Sciences, Botnar Research Centre, Oxford OX3 7LD, UK
British Society of Paediatric and Adolescent Rheumatology (BSPAR) Study Group:
M Abinum, MD, M Becker, MD, A Bell, MD, A Craft, MD, E Crawley, MD, J David, MD, H Foster, MD, J Gardener-Medwin, MD, J Griffin, MD, A Hall, MD, M Hall, MD, A Herrick, MD, P Hollingworth, MD, L Holt, MD, S Jones, MD, G Pountain, MD, C Ryder, MD, T Southwood, MD, I Stewart, MD, H Venning. L Wedderburn, MD, P Woo, MD and S Wyatt, MD.
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Licence. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Hinks, A., Martin, P., Flynn, E. et al. Association of the CCR5 gene with juvenile idiopathic arthritis. Genes Immun 11, 584–589 (2010). https://doi.org/10.1038/gene.2010.25
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gene.2010.25
Keywords
This article is cited by
-
A bivalent compound targeting CCR5 and the mu opioid receptor treats inflammatory arthritis pain in mice without inducing pharmacologic tolerance
Arthritis Research & Therapy (2018)
-
Hartung-Knapp-Sidik-Jonkman approach and its modification for random-effects meta-analysis with few studies
BMC Medical Research Methodology (2015)
-
Independent replication analysis of genetic loci with previous evidence of association with juvenile idiopathic arthritis
Pediatric Rheumatology (2013)
-
Meta-analysis confirms association between TNFA- G238A variant and JIA, and between PTPN22-C1858T variant and oligoarticular, RF-polyarticular and RF-positive polyarticular JIA
Pediatric Rheumatology (2013)
-
Dense genotyping of immune-related disease regions identifies 14 new susceptibility loci for juvenile idiopathic arthritis
Nature Genetics (2013)
