
Abstract
Introduction
Oxidative damage of the retinal pigment epithelium (RPE) may play a role in the development and progression of age-related macula degeneration (ARMD). Therapeutic reduction of oxidative stress failed or had only slight effects in ARMD patients. This study evaluates antiapoptotic properties of erythropoietin (epo) at the RPE as a novel approach to protect RPE cells against oxidative damage.
Materials and Methods
Cultured ARPE-19 cells were exposed to hydroxyl (OH) radicals generated from H2O2 under catalysis of Fe3+ (Fenton reaction) for 5 min. Apoptosis rate was determined by Annexin V labelling and terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling assay. Epo was added in concentrations from 0 to 100 U/ml to the media 24 and 1 h before radical exposure as well as shortly after radical exposure. Expression of epo receptor was determined by western blotting.
Results
Hydroxyl radical exposure induced an increase of apoptosis rate from virtually 0 to 11.8±1.7%. Apoptosis was detectable up to 24 h after radical exposure and reached its maximum after 6 h. Epo reduced apoptosis rate by up to 88% even if applied after the radical exposure. Best protection was achieved at 5 U/ml epo. Western blot confirmed presence of epo receptor independent of a pre-incubation of the cells with epo.
Discussion
Epo exerts antiapoptotic effects on cultured RPE cells even if applied after the radical exposure. This might qualify epo as future candidate for therapy and prevention of dry ARMD.
Similar content being viewed by others


Introduction
Altered retinal pigment epithelium (RPE) function and apoptosis is thought to play a key role in the development of age-related macula degeneration (ARMD).1 There are many indications that oxidative stress contributes to the ARMD-associated RPE changes.2 The RPE has, even under normal conditions, a particular pronounced risk for oxidative damage due to its location in a highly oxygenated environment close to the choroid and its exposure to high levels of focused visible light.1 In ARMD, susceptibility of RPE to light—particularly to short-wavelength blue light—and subsequent oxidative damage is potentiated by lipofuscin in drusen and RPE cells, as lipofuscin contains fluorophores, such as A2E, which promotes the generation of radicals under the influence of blue light.3, 4 Thus, there exists a vicious circle of light exposure, lipofuscin accumulation and oxidative stress, which should be interrupted to stop ARMD progression.5 Removal of lipofuscin is impossible and filtering blue light by specific glasses or by blue filter intraocular lenses were tried but have not brought a breakthrough. In addition, numerous trials were performed to investigate effectiveness of antioxidative therapy. These clinical studies showed a large variability in their results and were able to show only slight beneficial effects in certain sub-populations of ARMD patients.6 An alternative idea would be to target the downstream cellular effects of oxidative stress, for example, apoptosis. In this study, it is hypothesised that erythropoietin (epo) prevents radical-induced apoptosis in RPE cells. Although antiapoptotic and trophic properties of epo have been shown in several tissues, including the brain and the retina, no data are available regarding the epo effect on the RPE.7, 8, 9, 10
Materials and methods
Cell culture
ARPE-19 cells, a spontaneously proliferating human RPE cell line, were seeded in 24-well plates and cultured in complete culture medium consisting of DMEM/F12 (1 : 1) medium (GIBCO, Invitrogen, Grand Island, NY, USA), containing 10% fetal bovine serum, glutamine, pencillin/streptomycin, insulin-transferrin-selenite (Roche), non-essential amino acids and HEPES buffer (1M), and incubated in tissue culture incubator with humidified atmosphere of 5%CO2 and 95% air at 37°C.
Radical exposure
Radical exposure was done with confluent ARPE-19 cell cultures. Cell culture medium was exchanged for a Krebs–Henseleit buffer (118 mM NaCl, 5 mM KCl, 1.2 mM MgCl2, 1.2 mM Na2SO4, 2 mM NaH2PO4) 1 h before radical exposure. This was done to eliminate the potential antioxidative effects of components of the medium. Hydroxyl radicals (OH radicals) were generated by the Fenton reaction from 0.75 mM H2O2 and 10 μM of the iron redox chelate Fe3+/nitrilotriacetate (Fe-NTA):


The sum reaction of (1) and (2) is the Fenton reaction:

The infusion of OH radicals into the plates was performed as described elsewhere.11 Briefly, a specifically designed superfusion cell culture system was used to ensure stable radical concentrations during the radical exposure, which lasted 5 min in the presented experiments. This system was evaluated by NMR technology and was shown to generate OH-radical concentrations in pathophysiological relevant concentrations.12 According to this work, 0.75 mM H2O2 and 10 μM Fe-NTA were taken as reference concentration. To increase the radical dosage H2O2 and Fe-NTA concentrations were increased proportional. In the manuscript, concentrations of radicals are expressed as multiples of the reference concentration. After withdrawal of the radicals, the cell culture was set back to the normal media. Apoptotic cell death was monitored up to 24 h after OH-radical exposure.
Protective effect of recombinant human epo
A quantity of 40 000 U/ml of recombinant human Epo (rhEpo; gift of Janssen-Cilag, Neuss, Germany) were diluted to 10 U/μl with sterile 150 mM saline solution, aliquoted, and then kept in a refrigerator. The ARPE-19 cell culture medium was replaced by rhEpo-containing medium at final concentrations of 0, 1, 5, 10, 100 U/ml, respectively for 24 h before exposure to OH radicals. For post-treatment, the cultures were treated with 0.1, 1, 5, 10, 100 IU/ml immediately after the application of OH radicals.
Labelling of apoptotic cells by Annexin V-FITC
As externalisation of phosphotidylserine occurs in the earlier stages of apoptosis, Annexin V-FITC staining can detect apoptosis at early stages. The membranes of RPE-19 cells undergoing apoptosis were identified using ApoAlert Annexin V assay (Clontech, CA, USA), according to the manufacturer's instructions. Briefly, after treatment adherent cells were washed twice with PBS and then rinsed once with binding buffer. The cells were then incubated with Annexin V diluted in the binding buffer for 15 min in the dark at room temperature. A CytoFluor 4000 Multiwell Plate Reader (PerSeptive Biosystems) quantified fluorescence of living cells, and data collection were accomplished with the CytoFluor Fluorescence Reader Program (Version 3.4, PerSeptive Biosystems). The cells were excited at wavelength of 485 nm and emission was recorded at 530 nm. Emission at 530 nm increases proportional to the number of apoptotic cells. Apoptosis rate was calculated by normalisation to the fluorescence of untreated control cell ARPE-19 cultures.
TUNEL assay of nuclear apoptosis
Visualisation and quantification of apoptotic cells was carried out by labelling of 3′-OH ends of DNA fragments by terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling (TUNEL) assay using the In situ Cell Death Detection Kit, Fluoresceine (Roche/Boehringer, Mannheim, Germany). Briefly, cells were exposed to reactive oxygen species, fixed with 4% paraformaldehyde for 1 h and stained according to the manufacturer's protocol. Fluorescence of fluoresceine-DNA complex is inversely proportional to the degree of DNA fragmentation in cells undergoing apoptosis. Cell fluorescence was measured using CytoFluor 4000 fluorescent plate reader at excitation/emission wavelengths of 485/530. Fluorescence of living cells was determined and normalised to the fluorescence of normal cells in the fellow control experiments. Background fluorescence was measured on each plate and subtracted. Each data point was the mean value of fluorescence readings of three independent wells. Apoptosis rate was calculated as percentage of fluorescence of cells treated with OH radicals to fluorescence of untreated control sample. No TdT enzyme was added to determine background fluorescence.
Western blot analysis of epo receptor
Cells were grown on 75 cm2 tissue culture flasks and at confluency (∼7 × 106) the medium was changed to another flask containing 1 U/ml of rhEpo and incubated at 37°C for 10 min. On ice, medium was removed and the cells were washed twice with ice-cold PBS and then lysed by adding 500 μl of 10 mM Tris-HCl (pH 7.5)/150 mM NaCl/5 mM EDTA/40 mM NaF/1 mM Na3VO4/1% Nonidet P-40/1% sodium deoxycholate/0.1% SDS/10 μM leupeptin/1 mM PMSF, and complete protease inhibitor. The adherent cells were scraped off from the flask using cold plastic scraper and then the cells were incubated on ice for 30 min. The cell lysate was collected after centrifugation for 30 min at 14 000 r.p.m. The protein concentration was measured by BCA Protein Assay Reagent Kit (Pierce, Rockford, IL, USA). The cell lysate (25 μg) was boiled in Laemmli buffer and electrophoresed under reducing conditions on 12% polyacrylamide gels. Proteins were then transferred to a polyvinylidene difluoride membrane (Bio-Rad, Hercules, CA, USA). Membranes were blocked in 5% non-fat dried milk for 1 h, washed in 20 mM Tris-buffered saline (pH 7.5) containing 0.05% Tween-20 (TBST) and probed with polyclonal rabbit antihuman Epo-R Ig (Santa Cruz Biotechnology, 1 : 200 dilution) overnight at 4°C. Membranes were washed with TBST and then incubated for 1 h at room temperature with horseradish peroxidase conjugated polyclonal goat anti-rabbit Ig (Dako, 1 : 1000 dilution), followed by washing three times with TBST. The antigen Epo-R was detected using Immobilon Western Chemiluminescent Substrate (Millipore, Bedford, MA, USA) and visualised with ChemiImager 5500 (Alpha Innotech, CA, USA).
Statistics
All data are shown as mean±SD unless stated otherwise. Statistic comparisons were done by using an ANOVA or in case of paired comparison of two groups a paired t-test. P<0.05 was considered to be significant.
Results
Apoptosis induction by oxidative stress
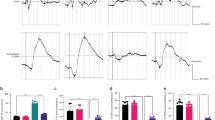
In the first set of experiments, induction of apoptosis by a transient 5 min lasting exposure to OH radicals was studied by Annexin V labelling. There was a concentration- and time-dependent effect of OH radicals on apoptosis rate. The maximum apoptosis rate was achieved 6 h after radical exposure. While the 1 × concentration did not exert any detectable increase in apoptosis rate, the maximum of apoptosis rate (11.8±1.7%; n=3) was found at the 3 × concentration after 6 h. With respect to this result, all further experiments were carried out with a 6 h follow-up (details and statistics in Figure 1).

Apoptosis rate (Annexin V labelling) in dependency of the radical concentration and follow-up time. The maximum apoptosis rate was reached 6 h after radical exposure with the threefold radical concentration. Each column represents n=3 experiments. Asterisk (*) denotes statistical significance (P<0.05).
The same apoptosis rate in dependence on radical concentration was observed using TUNEL labelling (Figure 2). Comparable to the values obtained after 6 h using Annexin V labelling, the TUNEL labelling showed after 6 h at the 3 × radical concentration an apoptosis rate of 12.2±1.5% (n=3).

Confirmative experiments with TUNEL assay (n=3 per group). Follow-up time was 6 h. Asterisk (*) denotes statistical significance.
Apoptosis inhibition by epo
To show that epo should be in general able to stimulate RPE cells, the presence of the epo receptor was confirmed by western blotting (Figure 3). To test the protective effect of epo on RPE cells, two application protocols were used. In one protocol, the cell culture was pre-incubated with epo for 24 or 1 h before radical exposure. In the second protocol, epo was applied shortly after the OH-radical treatment. Epo concentrations between 1 and 100 U ml−1 were used. The apoptosis rate after OH-radical exposure is clearly decreased by epo in both application protocols. The maximum reduction of apoptosis was 88% and was achieved at 5 U ml−1. Interestingly, at higher concentrations the protective effect of epo was reduced when the cells were pre-incubated with epo, but not when epo was applied after the OH-radical exposure. Data and statistics are summarised in Figure 4.

Western blotting of the erythropoietin receptor (EpoR) of ARPE-19 cells (1) and ARPE-19 cells that were pre-incubated for 24 h with recombinant human erythropoietin (2). EpoR is expressed in ARPE-19 cells and seems not to be affected by Epo pre-incubation.

Effect of recombinant human erythropoietin (rhEpo) on apoptosis rate after radical exposure. rhEpo showed antiapoptotic effects in ARPE-19 cells and was even effective if applied shortly after radical exposure. The protective effect gets lost if the cells were pre-incubated with higher concentrations of rhEpo (n=3 per group; *P<0.05).
Discussion
This study confirms the hypothesis that epo can prevent apoptosis induced by oxygen-free radicals. Astonishingly, epo was effective even when applied after the cellular intoxication with OH radicals. This makes it particularly attractive for therapeutic applications. Compared with the antioxidative approaches, the protection provided by epo to the RPE appears to be more efficient.11
Most of the earlier attempts to protect RPE cells from free radical-induced injury were based on scavenging the free radicals. The results of these attempts could not be confirmed in laboratory investigations and clinical trials. Ascorbic acid, for example, has in conjunction with oxidative stress toxic effects on RPE cells at low, more physiologically, or pharmacologically relevant concentrations.11 Pyruvate, another potent oxygen-free radical scavenger, shows similar properties.11 In clinical studies, the AREDS trial showed only slight beneficial effects of an antioxidative therapy in some ARMD sub-populations.6 These differences observed in antioxidative therapies are discussed to result from an uncontrolled intervention in the antioxidative defence network of the cells.13
In this study, it could be shown that RPE cells express the epo receptor and that stimulation of RPE cells with epo either before radical exposure or shortly after radical exposure could remarkably reduce apoptosis rates. However, this study cannot answer the question how epo acts in RPE cells. The fact, that epo is still efficient when applied after the radical exposure implies a specific cellular mechanism that does not involve a simple scavenging effect of the epo molecule. As RPE cells according to the results of this study express the epo receptor, the receptor might be most likely the starting point for the signal-transduction pathway, which is responsible for the epo effect. Future studies will be necessary to characterise the underlying cellular signal transduction in RPE cells. In other tissues the antiapoptotic effect of epo is reported to be mediated by activation of the Janus Tyrosine Kinase 2 (JAK-2), which phosphorylates proteins of the signal transducer and transcription (STAT) family.14, 15 Particularly STAT5 is thought to play an important role in stimulating antiapoptotic proteins such as Bcl-XL and Bcl-2.16 JAK-2 activates beside STAT5 NF-kB, which contributes to neuroprotection in neurons. Although neither the JAK-2/STAT nor the NF-kB pathway is investigated systematically in RPE cells, there are some data published indicating a JAK-2 activation by epo in RPE cells, which protects cells against light damage.17 The authors would suggest to take an involvement of these pathways into account for conception of future work on epo at the RPE.
Interestingly, the protective effect of epo gets lost at higher concentrations of epo, but only if applied before radical exposure and not if applied after radical exposure. There is no conclusive explanation for this observation up to now. In BOSC-23T cells, it was shown that stimulation of the epo receptor results in a downregulation of the epo-mediated signalling cascade by dephosphorylation of the epo receptor by protein tyrosine phosphateases.18 Such a mechanism might also be present at the RPE, but has not been investigated yet. Thus, one might speculate that higher concentrations of epo lead in the present experiments to modifications at the receptor or in the downstream signalling cascade, which have an inactivating effect on the antiapoptotic processes controlled by epo.
Like in this study at the RPE, epo has been identified as a trophic factor on other tissues, including kidney, brain, and retina, in the past.7, 9, 10 This has led to the consideration of epo in the therapy of stroke patients with the aim to reduce the collateral neuronal damage.19 It was furthermore shown that epo preserves cognitive function in schizophrenic patients.20 In experimental models of retinal degeneration epo has been shown to provide successful protection.8 A recent study by a Chinese group indicates in accordance with the present work protective properties of epo at light-exposed RPE cells.17 This study and the present results show a rationale for further experiments investigating the trophic and protective effects of epo on the RPE in vivo.
Of course, a direct application of the presented results of this in vitro study to the in vivo situation has to be done with care. Although ARPE-19 cells are used as a kind of standard cell culture model for RPE and it was shown that several features of the primary RPE cells are preserved in this cell line, RPE in vivo might behave differently.1
Future research should be focused on the identification of potential fields of therapeutic indications for epo. The first step will be a systematic evaluation of epo in different animal models, for example, for ARMD, ocular vascular diseases, and retinal degeneration. The authors feel that dry ARMD would be one of the most challenging and promising fields as primary apoptosis of RPE cells play a crucial role in the pathogenesis of this disease. Special attention has to be paid to angiogenic properties of epo.21, 22 In conjunction with ARMD this might promote the switch from a dry to a wet ARMD. Furthermore, the dependency of the epo effect from the time point of administration arises the need to characterise the critical time point for epo administration more precise to figure out optimal treatment conditions. In addition, the mode of administration has to be investigated. There are no data available on the bioavailability of systemically given epo at the retina or the RPE. In times of large-scale intravitreal injections, it appears to be obvious to apply epo intravitreally, which allows targeting at quite defined intraocular concentrations. Recently, it has been shown in animal experiments that intravitreal injections of epo are safe and efficient.23, 24
In summary, this study introduces a new approach how to protect RPE cells against the effects of oxidative stress without touching the delicate antioxidative defence network of the cells.
References
Strauss O . The retinal pigment epithelium in visual function. Physiol Rev 2005; 85 (3): 845–881.
Beatty S, Koh H, Phil M, Henson D, Boulton M . The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol 2000; 45 (2): 115–134.
Sparrow JR, Zhou J, Ben-Shabat S, Vollmer H, Itagaki Y, Nakanishi K . Involvement of oxidative mechanisms in blue-light-induced damage to A2E-laden RPE. Invest Ophthalmol Vis Sci 2002; 43 (4): 1222–1227.
Rozanowska M, Jarvis-Evans J, Korytowski W, Boulton ME, Burke JM, Sarna T . Blue light-induced reactivity of retinal age pigment. In vitro generation of oxygen-reactive species. J Biol Chem 1995; 270 (32): 18825–18830.
Rozanowska M, Wessels J, Boulton M, Burke JM, Rodgers MA, Truscott TG et al. Blue light-induced singlet oxygen generation by retinal lipofuscin in non-polar media. Free Radic Biol Med 1998; 24 (7–8): 1107–1112.
The AREDS Investigator Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol 2001; 119 (10): 1417–1436.
Marti HH . Erythropoietin and the hypoxic brain. J Exp Biol 2004; 207 (Part 18): 3233–3242.
Grimm C, Wenzel A, Groszer M, Mayser H, Seeliger M, Samardzija M et al. HIF-1-induced erythropoietin in the hypoxic retina protects against light-induced retinal degeneration. Nat Med 2002; 8 (7): 718–724.
Weishaupt JH, Rohde G, Pölking E, Siren AL, Ehrenreich H, Bähr M . Effect of erythropoietin axotomy-induced apoptosis in rat retinal ganglion cells. Invest Ophthalmol Vis Sci 2004; 45 (5): 1514–1522.
Junk AK, Mammis A, Savitz SI, Singh M, Roth S, Malhotra S et al. Erythropoietin administration protects retinal neurons from acute ischemia-reperfusion injury. Proc Natl Acad Sci USA 2002; 99 (16): 10659–10664.
Zeitz O, Schlichting L, Richard G, Strauss O . Lack of antioxidative properties of vitamin C and pyruvate in cultured retinal pigment epithelial cells. Graefes Arch Clin Exp Ophthalmol 2007; 245 (2): 276–281.
Zweier JL, Thompson-Gorman S, Kuppusamy P . Measurement of oxygen concentrations in the intact beating heart using electron paramagnetic resonance spectroscopy: a technique for measuring oxygen concentrations in situ. J Bioener Biomembr 1991; 23 (6): 855–871.
Chen Q, Espey MG, Krishna MC, Mitchell JB, Corpe CP, Buettner GR et al. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: action as a pro-drug to deliver hydrogen peroxide to tissues. Proc Natl Acad Sci USA 2005; 102 (38): 13604–13609.
Ihle JN . Cytokine receptor signalling. Nature 1995; 377 (6550): 591–594.
Siren AL, Fratelli M, Brines M, Goemans C, Casagrande S, Lewczuk P et al. Erythropoietin prevents neuronal apoptosis after cerebral ischemia and metabolic stress. Proc Natl Acad Sci USA 2001; 98 (7): 4044–4049.
Silva M, Benito A, Sanz C, Prosper F, Ekhterae D, Nuñez G et al. Erythropoietin can induce the expression of bcl-x(L) through Stat5 in erythropoietin-dependent progenitor cell lines. J Biol Chem 1999; 274 (32): 22165–22169.
Meng Y, Niu YJ, Qu H . [The research of the protection of recombinant human erythropoietin in human RPE cells by light-induced injuries]. Zhonghua Yan Ke Za Zhi 2008; 44 (1): 50–55.
Cohen J, Oren-Young L, Klingmuller U, Neumann D . Protein tyrosine phosphatase 1B participates in the down-regulation of erythropoietin receptor signalling. Biochem J 2004; 377 (Part 2): 517–524.
Ehrenreich H, Hasselblatt M, Dembowski C, Cepek L, Lewczuk P, Stiefel M et al. Erythropoietin therapy for acute stroke is both safe and beneficial. Mol Med (Cambridge, Mass) 2002; 8 (8): 495–505.
Ehrenreich H, Hinze-Selch D, Stawicki S, Aust C, Knolle-Veentjer S, Wilms S et al. Improvement of cognitive functions in chronic schizophrenic patients by recombinant human erythropoietin. Mol Psychiatry 2007; 12 (2): 206–220.
Watanabe D, Suzuma K, Matsui S, Kurimoto M, Kiryu J, Kita M et al. Erythropoietin as a retinal angiogenic factor in proliferative diabetic retinopathy. N Engl J Med 2005; 353 (8): 782–792.
Jaquet K, Krause K, Tawakol-Khodai M, Geidel S, Kuck KH . Erythropoietin and VEGF exhibit equal angiogenic potential. Microvasc Res 2002; 64 (2): 326–333.
Song BJ, Cai H, Tsai JC, Chang S, Forbes M, Del Priore LV . Intravitreal recombinant human erythropoietin: a safety study in rabbits. Curr Eye Res 2008; 33 (9): 750–760.
Zhang J, Wu Y, Jin Y, Ji F, Sinclair SH, Luo Y et al. Intravitreal injection of erythropoietin protects both retinal vascular and neuronal cells in early diabetes. Invest Ophthalmol Vis Sci 2008; 49 (2): 732–742.
Acknowledgements
This study was supported by the Grant nos. 341 and 351 from the Claere Jung Stiftung, Hamburg, to Oliver Zeitz.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gawad, A., Schlichting, L., Strauß, O. et al. Antiapoptotic properties of erythropoietin: novel strategies for protection of retinal pigment epithelial cells. Eye 23, 2245–2250 (2009). https://doi.org/10.1038/eye.2008.398
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2008.398
