
Abstract
The endoplasmic reticulum (ER) is an important subcellular organelle that is involved in numerous activities required to achieve and maintain functional proteins in addition to its role in the biosynthesis of lipids and as a repository of intracellular Ca2+. The inability of the ER to cope with protein folding beyond its capacity causes disturbances that evoke ER stress. Cells possess molecular mechanisms aimed at clearing unwanted cargo from the ER lumen as an adaptive response, but failing to do so navigates the system towards cell death. This systemic approach is called the unfolded protein response. Aging insults cells through various perturbations in homeostasis that involve curtailing ER function by mitigating the expression of its resident chaperones and enzymes. Here the unfolded protein response (UPR) cannot protect the cell due to the weakening of its protective arm, which exacerbates imbalanced homeostasis. Aging predisposed breast malignancy activates the UPR, but tumor cells maneuver the mechanistic details of the UPR, favoring tumorigenesis and thereby eliciting a treacherous condition. Tumor cells exploit UPR pathways via crosstalk involving various signaling cascades that usher tumor cells to immortality. This review aims to present a collection of data that can delineate the missing links of molecular signatures between aging and breast cancer.
Similar content being viewed by others
Introduction
Breast cancer, like other cancers, results from interactions of various factors, such as genetic predisposition and the environment. The scientific repertoire has an enormous amount of data on the mechanisms of breast cancer development, its diagnosis, prevention and treatment. The most important aspect of any cancer study is the risk factors. When we discuss breast cancer, among all other risk factors, getting older, that is, aging, is known to be a condition that promotes the disease. The American Cancer Society has previously shown that most invasive breast cancer cases are reported among women aged 55 years or more.1 Hence, there is evidence suggesting that malignancies are a direct function of aging.2, 3 Although aging accompanies the degeneration and loss of function of various tissues in the body, especially the skeletal muscles, the development of a tumor and its progression in aging is blamed on the accumulated mutations in oncogenes and tumor suppressors.4
Breast cancer has multiple molecular classifications: luminal A and B, triple-negative/basal-like (TNBC; tumors that do not express genes for the estrogen receptor, progesterone receptor and human epidermal growth factor receptor 2 (HER2)) and HER2-positive subtypes.5 Aging does lead to changes in the expression pattern of the transcriptome, as shown by previous studies in various models of breast cancer.6, 7, 8, 9 The field of geroscience is still in its infancy and is evolving to understand the paradigm of breast cancer.
Environmental factors constantly affect the cellular microenvironment. There are a number of adverse effects posed by environmental factors, such as radiation and mutagens, that lead to intracellular damage, whereby cellular deterioration substantially contributes to aging and tumorigenesis. This damage primarily involves important components of cells: the proteins in their proper folded, functional form. Under normal conditions, the endoplasmic reticulum (ER) controls the damaged/misfolded protein overload by activating rescue pathways that upregulate the expression of resident chaperones to properly fold the misfolded proteins, halting its translational function and appropriately diverting the unwanted cargo towards a degradation pathway, the endoplasmic reticulum-associated degradation (ERAD).10
The overall aim is to restore a healthy cellular microenvironment, directing the cell towards homeostasis. The overload of misfolded and accumulated proteins invariably disturbs the quality control process in the ER lumen, thereby presenting a condition of ‘ER stress.’11 The molecular machinery in the ER lumen tackles this stress via a multifarious response cascade called the unfolded protein response (UPR). The UPR is an orchestration of complex events comprising an increased rate of protein folding by upregulating resident molecular chaperones and the diversion of the extra misfolded proteins to degradation pathways aimed at rescuing the cell from the imposed stress.
Onset of the endoplasmic reticulum unfolded protein response
The ER lumen harbors important molecular chaperones, folding sensors and enzymes that are engaged in protein quality control, that is, proper folding of nascent polypeptides. These include glucose regulated proteins 78 (GRP78)/immunoglobulin-binding protein, GRP94, calnexin, calreticulin, protein disulfide isomerase, thiol-disulfide oxidoreductase and ERp57.12, 13 Studies have shown that aging leads to a condition in which there is downregulation in the expression pattern of resident molecular chaperones and enzymes in the ER across several tissues.14 This in turn lowers the folding efficiency of the ER, thereby increasing the bulk of misfolded proteins. This instability in protein homeostasis results in ER stress that consequently stimulates the UPR.15 Stressors arise from the tumor microenvironment, such as diminishing levels of available oxygen and nutrient supplies, leading to UPR activation in malignant cells.16, 17, 18
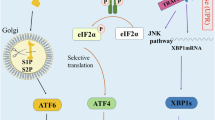
UPR signaling involves three resident transmembrane ER proteins, PERK (the PKR-like ER kinase), IRE-1 (the inositol requiring element 1) and ATF-6 (the activating transcription factor 6), which act in a complex multifaceted cascade aimed at restoring proteostasis in the ER lumen. A diagrammatic representation of the events showing activation of UPR transducers and the subsequent downstream signaling effectors is presented in Figure 1. Activation of PERK and IRE-1 is marked by their homo-dimerization followed by trans-autophosphorylation of their cytoplasmic components, while ATF-6 activation leads to its translocation to the Golgi apparatus where two serine proteases (site-1 protease, S1P and site-2 protease, S2P) cleave it.

The unfolded protein response during ER stress. The molecular chaperon GRP78 is associated with the luminal components of ER membrane-resident UPR transducers, PERK, IRE-1 and ATF-6, thereby preventing their activation in a non-stressed cell. When the amount of misfolded proteins escalates in the ER lumen, GRP78 leaves the UPR transducers and is recruited for protein folding. Upon losing their association with GRP78, the transmembrane UPR transducers undergo changes, an event marking the activation of the UPR. The GRP78-free PERK homodimerizes and undergoes trans-autophosphorylation at its cytoplasmic component. This event targets the phosphorylation of cytoplasmic eIF2α thereby affecting the UPR through the PERK arm, leading to the attenuation of general translation in the cell. However, this event does not affect the translation of ATF-4 mRNA through its internal ribosome-binding site element. ATF-4 translocates into the nucleus where it acts on CHOP genes, leading to the expression of genes required for amino-acid metabolism, antioxidant response and apoptosis. ATF-4 also induces the expression of CHOP that potentiates the expression of genes responsible for cell death. When the IRE-1 arm loses its association with GRP78, it also dimerizes and trans-autophosphorylates, activating its RNase activity. The endoribonuclease property of IRE-1 leads to unconventional splicing of XBP-1 mRNA. The spliced variant is a 26 bp RNA segment called XBP-1(S), which is translated into the XBP-1 protein that is translocated into the nucleus to upregulate the expression of genes involved in ER expansion, protein maturation and the regulation of cargo protein secretion out of the ER lumen. ATF-6, which is a 90 kDa transmembrane protein, translocates to the Golgi apparatus membrane after GRP78 leaves its luminal component. The action of two serine proteases, S1P and S2P, releases a 50 kDa cytosolic fragment, p50ATF-6, which translocates into the nucleus to upregulate the expression of ER molecular chaperones and enzymes.
GRP78 has a vital role in protecting the cell during ER stress. During normal cell functions, GRP78 is associated with the luminal domains of the UPR transducers. The association of GRP78 sterically prevents the homo-dimerization of PERK and IRE-1 and also inhibits the translocation of ATF-6 to the Golgi apparatus. When the concentration of misfolded proteins increases in the ER lumen, GRP78 disassociates from the transmembrane stress transducers and is recruited to protein folding, a marked step that signals ER stress in the cell. Apart from this, the GRP78 prevents cell death triggered by cytosolic accumulation of Ca2+ during ER stress by withholding luminal Ca2+ in the ER, thereby preventing its leakage into the cytosol.19
ERAD focuses on diverting the aberrant and misfolded protein cargo towards a systematic degradation pathway. The process invariably starts with tagging unwanted bulk misfolded proteins with molecular chaperones and their subsequent retro-translocation back into the cytosol, where poly-ubiquitination ensues ending in 26S proteasomal degradation.10 Thus, ERAD functions to direct the cell towards adaptive UPR.20, 21 Autophagy is another process that clears the misfolded protein accumulation from the cell.22
After the occurrence of ER stress in the cell, UPR signaling first triggers an adaptive cascade through programming of a series of events, which are pro-survival. But if the stress condition is not alleviated, UPR commits to the activation of a pro-death cascade.
PERK-stimulated phosphorylation of eIF2α and the attenuation of cellular mRNA translation
During the UPR, GRP78 detached PERK gets homodimerizes and transduces the signal through autophosphorylation of its cytoplasmic domain. This dissipates its effect, causing phosphorylation of cytoplasmic eukaryotic initiation factor alpha (eIF2α), whereby the global protein synthesis comes to an immediate halt.23 The phosphorylated eIF2α lowers the levels of the active eIF2α-GTP pivotal for the association of Met-tRNAiMet with the 40S ribosome in the formation of the ternary complex of the translational apparatus.24, 25 This action is aimed at reducing the amount of fresh proteins in the ER lumen. However, this does not stop the translation of certain selected proteins, such as activating transcription factor-4 (ATF-4) and GRP78 by exploiting their internal ribosome-entry site (IRES) elements in mRNA.26, 27 The human and mice ATF-4 mRNA are characterized by two upstream open reading frames (uORFs) in the 5′ non-coding region of the transcript. uORF1 codes for a short 3 amino-acid tripeptide, whereas the uORF2, which overlaps the first 83 nucleotides of the ATF-4 coding region, encodes a polypeptide with 59 amino acids.28, 29 In normal cells, eIF2-GTP-Met-tRNAiMet bound to the 40S ribosome machinery starts its scan from the 5′-end of ATF-4 mRNA and translates the uORF1, exerting a positive effect by facilitating the retention of the translational apparatus for immediate reinitiation of the upcoming uORF2. The translational machinery scans past the overlapping, now out-of-frame AUG of ATF-4 ORF in uORF2, impeding ATF-4 translation. While during ER stress, under diminishing levels of eIF2α–GTP due to phosphorylated eIF2α, the 40S translational machinery spends more time on the reinitiation of translation of uORF2; this delayed time interval selects for the translation of ATF-4 ORF (delayed reinitiation model).29
This bypassed translational upregulation of ATF-4 is called the integrated stress response (ISR).30 ATF-4 is a transcription factor that controls the expression of genes involved in amino-acid metabolism, the antioxidant response and apoptosis.28, 30, 31 The transcription of the C/EBP (CCAAT/enhancer-binding protein) homologous protein, CHOP (also known as growth arrest and DNA damage 153, GADD153), is activated by ATF-4, which is pro-death.26, 32, 33, 34
IRE-1-induced unconventional splicing of XBP-1
The second transducer of ER stress, IRE-1, upon losing its association with GRP78, dimerizes and trans-autophosphorylates, marking IRE-1 activation. The activated IRE-1 has RNase activity. The substrate for this endoribonuclease is the intron of mRNA that codes for X-box-binding protein-1 (XBP-1), a transcription factor specific to the UPR. This unconventional splicing removes the 26 bp nucleotide, yielding a spliced variant called XBP-1(S), which can alleviate ER stress by acting as a transcriptional activator of genes involved in ER expansion, protein maturation and secretion, as well as clearing the misfolded protein overload via degradation.35, 36 Cells overexpressing XBP-1 inhibit CHOP expression, which is pro-survival.37 However, there are compelling reports that challenge this idea of IRE-1-XBP-1 being pro-survival during acute ER stress.38 Additionally, reports indicate that IRE-1 binds directly to unfolded proteins, thereby acting as an activating ligand itself.39, 40
ATF-6 translocation to the Golgi and its activation
The third transmembrane sensor, ATF-6, is a 90 kDa protein. Upon disassociating from GRP78, as a part of regulated intramembrane proteolysis, this ER membrane-resident transcription factor translocates to the Golgi apparatus, where it is cleaved by serine proteases (site-1 protease, S1P and site-2 protease, S2P), releasing a 50 kDa cytosolic fragment called p50ATF-6, the functional isoform.41 This transcription factor is imported into the nucleus where it targets the cis-acting ER stress response elements.42 ATF-6 controls the expression of genes coding for ER-resident molecular chaperones and enzymes involved in protein folding.43, 44 Most importantly, ATF-6 accentuates the expression of GRP78 to ameliorate the built-up stress.45 Studies confirm the interaction of XBP-1 and ATF-6 during UPR.44
ER stress-induced apoptosis
The orchestrated functioning of the three transmembrane ER stress transducers, PERK, IRE-1 and ATF-6, is aimed at mitigating the stress and the restoration of proteostasis, and thereby cellular homeostasis. However, prolonged stress, and burdening factors such as aging may lead the cell towards apoptosis.46 Two pathways govern the cellular control of apoptosis with molecular crosstalk: the intrinsic or mitochondrial pathway and the extrinsic or death receptor pathway. The stimuli for the onset of the intrinsic pathway, such as DNA damage, suppression of growth hormones and cytokines, lead to changes in the inner mitochondrial membrane causing the exposure of pro-apoptotic proteins.47 There is efflux of ER luminal Ca2+ into the cytosol, which dissipates its effect on caspase-12.48 In two separate studies, the activation of caspases is shown during ER stress-induced cell death. Nakagawa et al.49 demonstrated that mice lacking caspase-12 genes were partially resistant to pharmacological inducers of ER stress, such as tunicamycin and thapsigargin. Rao et al.50 demonstrated that caspase-7 may activate caspase-12 by translocating from the cytosol to the ER. The ER stress succumbing cell activates CHOP at various levels. PERK phosphorylates the alpha subunit of eIF2α but paradoxically activates ATF-4, which affects the CHOP promoter.51 CHOP is also a component of the downstream effectors of ATF-6. These factors collectively stimulate the expression of pro-apoptotic proteins such as growth arrest and DNA damage inducible protein 34 (GADD34), TRB3 (tribbles-related protein 3) and so on.33, 52 The activation of GADD34 by CHOP leads to ER luminal protein synthesis by dephosphorylation of eIF2α aimed at increasing protein load in the ER.33, 53 CHOP potentiates the upregulation of pro-apoptotic proteins belonging to the BCL2 family such as BAK/BAD and the downregulation of anti-apoptotic proteins, leading to the release of cytochrome c from the mitochondrial membrane into the cytosol, an event that further activates cytosolic apoptotic protease activating factor1 (APAF1). This further activates caspase-3- and caspase-9-dependent signaling.54, 55 CHOP is also positively regulated by p38 kinase.56
Extracellular stimuli, such as transmembrane receptor-mediated interactions, lead to the onset of the extrinsic pathway of apoptosis. Members of the tumor necrosis factor receptor gene superfamily, such as death receptors, are involved in this process.57, 58 Reports point to the activation of c-Jun N-terminal kinase (JNK), a family of signal transducers activated by exogenous stimuli, during ER stress.59 The tumor necrosis factor receptor-associated factor2 (TRAF2), which is bound to the cytoplasmic component of IRE-1 and transduces signals from IRE-1, activates JNK.59 TRAF2 also invariably promotes caspase-12 activation.59 Reports suggest that kinases, such as apoptotic signal regulating kinase-1, are activated, causing further stimulation of JNK and p38 MAPK.60
Activation of the inflammatory signaling cascade during the UPR
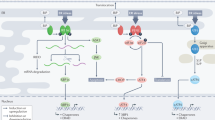
Increasing evidence suggests that the inflammatory signaling cascade responds to the cellular insults imposed during ER stress. The production of reactive oxygen species, leakage of ER luminal Ca2+ into the cytosol and the activation of nuclear factor-κB (NF-κB), the master transcriptional regulator of pro-inflammatory signaling, are some examples that point to the connection between the UPR and inflammation.31, 61, 62 A schematic of these events is presented in Figure 2 and described as follows. In a normally functioning cell, NF-κB is primarily maintained in its inactive form through binding with its constitutively expressed inhibitors, inhibitors of NF-κB (IκB). The imposed stress stimulates the activation of NF-κB, after which it translocates into the nucleus to upregulate inflammatory genes. The stressful condition in the ER lumen due to high oxidative stress created by increased protein folding and the leakage of luminal Ca2+ plays a role in NF-κB activation.63 Additionally, PERK-eIF2α-directed translation attenuation activates NF-κB. The half-life of IκB is low, so the translational attenuation frees NF-κB.64, 65

The relationship between the inflammatory signaling cascade and the UPR. The stress generated in the ER lumen by the accumulation of reactive oxygen species and the leakage of intraluminal Ca2+ leads to the activation of NF-κB, the master transcriptional regulator of pro-inflammatory signaling. The PERK-eIF2α-induced attenuation of cellular translation activates NF-κB. IRE-1 forms a complex with TRAF2 to activate NF-κB. This complex also activates JNK through activator protein 1, which further upregulates the expression of apoptotic genes. The p50ATF-6 fragment generated after intramembrane proteolysis of ATF-6 in the Golgi apparatus also activates NF-κB. Another candidate for the stimulation of the inflammatory response, mTOR, upregulates the expression of genes involved in ERAD and is activated through Rheb by p50ATF-6.
IRE-1 also integrates the UPR signal to an inflammatory response. NF-κB can also be activated after the formation of the IRE-1–TRAF2 complex during ER stress.65 Studies on IRE-1 knockdown in mouse embryonic fibroblasts support the activation of NF-κB by UPR and production of inflammatory cytokine tumor necrosis factor-α (TNFα).65 As mentioned previously, the IRE-1–TRAF2 complex also activates JNK, which upregulates the expression of inflammatory genes through the phosphorylated activator protein 1 (AP1).66 Studies on subtilase cytotoxin (SubAB), a toxin produced by Shiga toxigenic E. coli, show that ATF-6 also accentuates the NF-κB during UPR.67
The mammalian target of rapamycin (mTOR), a serine/threonine protein kinase of the phosphatidylinositol-3-OH kinase (PI(3)K)-related family, plays a pivotal role in the regulation of proliferation and metabolism in various cellular processes.68, 69, 70, 71, 72, 73 There are two multiprotein complexes of mTOR, mTOR complex 1 (mTORC1) and 2 (mTORC2).71 Akt, also known as protein kinase B or PKB, is an important molecule involved in the regulation of cellular processes and is suppressed by mTORC1, whereby apoptosis is induced through the IRE-1–JNK pathway of UPR.74 Reports suggest the hyperactivation of mTOR during the inflammatory response.75
The crosstalk between the UPR and autophagy
Autophagy pathways are highly inducible under stress conditions and are mediated by a series of well-coordinated genes, autophagy-related genes.76 ER stress-induced autophagy can be cytoprotective or cytotoxic.77, 78 The conserved recycling pathway that helps the cell to maintain its ‘quality control’ system represents the cytoprotective facet of autophagy,79 whereas stress-induced mitigation of autophagy has been implicated in several diseases, including cancer.80, 81
The process of autophagy is initiated with the formation of a double membrane structure, the autophagosome, which engulfs the targeted molecule in the cytosol destined for the lysosome (Figure 3). LC3, the microtubule-associated protein 1 light chain 3, existing as a soluble protein, LC3-I, conjugates with phosphatidylethanolamine and is transformed into membrane-associated LC3-II protein, which is recruited to autophagosomal membranes. This key step is followed by a series of events, leading to the fusion of autophagosomes with lysosomes possessing integral membrane proteins, the lysosome-associated membrane proteins (LAMPs),82, 83, 84, 85 which constitute a family represented by glycosylated type I transmembrane proteins. The members LAMP-1 and LAMP-2 are found in the lysosome and late endosomes,86 and they make up half of the total integral membrane protein in the lysosome and maintain its functional integrity.87 LAMP-3 is expressed only in specific cell types and is under temporal and spatial regulation.88 LAMP-3 invariably contains more sites for glycosylation than LAMP-1 and LAMP-2.89

Autophagy in the UPR. The commencement of autophagy is the result of an orchestration of genes called autophagy-related genes. The process of autophagy involves molecular crosstalk with UPR transducers. The PERK/eIF2α arm of the UPR induces the expression of autophagy-related genes through ATF-4. Additionally, the sustained activation of XBP-1, which is downstream of IRE-1, promotes the recruitment of soluble LC3-I to the membranous structure, the autophagosome. Here it is transformed into its membrane-associated form, LC3-II. This autophagosome complex conjugated with LC3-II engulfs the organelle/molecule, which is destined to be degraded. This complex fuses with lysosome-expressing integral membrane proteins, LAMPs (1,2,3), leading to the formation of the autolysosome complex.
Evidence favoring the involvement of the UPR in the regulation of autophagy is accumulating. Studies have shown the involvement of the PERK/eIF2α arm of the UPR, in inducing the transcription of essential autophagy genes, which focuses on cell survival under hypoxic conditions.90 Studies have reported the atypical localization of LAMP-2 on the cell surface during human diseases such as cancer.91 Also, the expression of LAMP-3 surges in tumor cells, which are metastatic in nature.92, 93 Mujcic et al.94 have shown that the PERK/eIF2α/ATF-4 arm of the UPR systemically accentuates LAMP-3 induction under hypoxic conditions. The sustained activation of IRE-1 downstream and spliced XBP-1 mRNA triggers autophagy through the marker LC3.95
Autophagy also intersects with the inflammatory responses in the cell during the UPR. NF-κB activation is terminated during the induction of autophagy.96, 97 Studies have corroborated the antagonistic relationship between mTOR and autophagy.72 Conditions of starvation and a lack of growth factors lead to the induction of autophagy in the cell by the inactivation of mTOR.98
Aging and the UPR
Resistance to stress decreases with aging. Maintenance of proteostasis in cells declines with the process of aging, leading to protein toxicity that can cause various age-related diseases and disorders. Aging causes weakening of the UPR and drives the mechanism towards cell death.99, 100, 101 Studies on aged mice have shown that a decline in protein folding due to the oxidation of molecular chaperones in the ER lumen results in increased proteotoxicity.102 Not only are the existing molecular chaperones destroyed, but the expression levels of GRP78 have also been reported to decline significantly.100 Aging also eclipses the ER stress transducers of the UPR. Paz Gavilan et al.101 showed using RT-PCR that the mRNA expression of PERK significantly diminishes in aged rats compared with younger rats. Aging-accelerated failure of protective responses by the tripartite arms of the UPR further exacerbates the condition by the activation of apoptotic effectors of the UPR. The levels of CHOP and pro-apoptotic caspase-12 are reported to increase in aged mice during sustained ER stress, steering the cell toward death.99, 101 Further studies support that the increase in CHOP expression sensitizes the cell to the ill effects of oxidation.46 Aging further shortens the threshold of apoptosis by activating the kinase-directed apoptotic pathway in ER stress-sensitized cells. JNK kinases are upregulated through the IRE-1 kinase activity involving apoptotic signal regulating kinase-1 (ASK1).103 Adler et al.104 reported the cis-regulatory motifs associated with aging through microarray-based analysis and found that NF-κB was the strongest candidate enforcing aging-related features. mTOR, a mediator of stress responses, extends lifespan when suppressed.105, 106 Autophagy works in coherence with the UPR in normal cells in which stress is overcome by clearing the aggregated cargo of misfolded proteins, but aging further deregulates the fusion of autophagosomes with lysosomes.107, 108 The inhibitory effect of mTOR on autophagy is well documented in the case of aging.80, 81 Aging-associated diseases show diminishing levels of the LC3 marker, that mark the enfeeblement of autophagy.109
A correlation between aging and a reduced UPR has been implicated in several age-related conditions such as diabetes and a number of neurodegenerative disorders.64, 100, 110, 111
Status of the UPR in breast tumorigenesis
Cells may endure various environmental and pathophysiological insults that can occasionally lead to the development and progression of malignancies. In a normal scenario, the immunity of a mammalian system tries to withstand such abuses and fights back to restore homeostasis. However, these actions can lead to cell transformation, resulting in malignancies. There is accumulating evidence supporting the role of the UPR in the development and progression of cancer cells. The conundrum behind activation of the UPR during tumorigenesis is compelling. The increased proliferation of tumor cells is a burden on nutrient and oxygen requirements. The tumor cells trigger angiogenesis, resulting in hypoxia and nutrient depletion.112, 113, 114, 115 Cancer cells extensively exploit the molecular machinery of the ER, leading to ER stress involving meticulous alterations of UPR transducers and effectors, so as to turn them immortal by preventing apoptosis. The ER-resident molecular chaperone GRP78 is overexpressed in cases of common malignancies such as breast cancers.116, 117
The physiology of the breast presents a paradigm in which ER stress and the ensuing UPR control the normal functioning of the system. During lactation, normally ER is under increased pressure for milk protein production, but the UPR arising due to this ER stress navigates the cell toward survival (the pro-survival response is activated). Also, during the normal menstrual cycle, the breast responds to various hormonal surges, thereby establishing proper homeostasis through the tripartite arms of the UPR. In cases of the initiation of breast malignancies, stressors such as hypoxia, nutrient deprivation, and cytotoxic and endocrine therapeutic interventions lead to the activation of a range of stress responses, including the UPR.
Role of PERK in breast malignancy
Regarding the tripartite arms of the UPR, Bobrovnikova-Marjon et al.118 reported in their work on breast cancer cells that PERK was necessary for tumor proliferation. Studies have shown that hypoxic parts of the tumor microenvironment activate the translational control program, the integrated stress response, which helps the malignant cells to adapt to the imposed stress of hypoxia. The integrated stress response target ATF-4 is highly expressed in the hypoxic cores of tumors,119 supporting tumor immortalization.119 Harding et al.31 suggested that the activation of the integrated stress response provokes a cohort of genes that defend the tumor cells against oxidative bursts, selectively inducing glutathione biosynthesis.
The nuclear factor erythroid-derived 2-related factor (NRF2), a cytoplasmic transcription factor, is a novel, second substrate for the PERK arm of the UPR, which is phosphorylated and subsequently imported into the nucleus to support cell survival as a protective answer to the UPR.120 Under normal circumstances, NRF2 remains in an inactive state in the cytosol through its association with the actin-binding protein, Kelch-like ECH-associated protein (Keap1).121 ER stress-induced PERK activation leads to the dissociation of this NRF2–Keap1 complex, thereby aiding the translocation of NRF2 into the nucleus to act on its target, antioxidant response element (ARE). This event activates pathways for antioxidants, detoxifying enzymes, trafficking proteins and degrading them to alleviate stress in the cell.31, 120, 122, 123 Cullinan et al.62 have shown that the expression of CHOP, which is activated through the PERK arm of the UPR favoring apoptosis, is attenuated in NRF2 overexpressing cells. These data demonstrate that the PERK arm is pro-survival in cells with the accrual of NRF2 during the UPR.124 NRF2 has also been reported to promote cell survival in cases of malignancies.125, 126 The work of Bobrovnikova-Marjon et al.118 supports the inactivation of NRF2 in cancer cells with PERK deletion. PERK regulates homeostasis in cancer cells through the prevention of oxidative DNA damage checkpoints. For therapeutic intervention of breast malignancies, loss of the PERK arm of the UPR can result in the suppression of tumor onset.118 Studies show that NRF2 plays a role in the survival of breast tumors under a limiting microenvironment via chemoresistance.127
In a separate study by Nagelkerke et al.,93 the PERK/ATF-4 arm of the UPR activates the expression of LAMP-3, which supports migrating breast malignancies. An analysis published by Sawada et al.128 corroborates that this migration characteristic of malignant cells is due to the overexpression of LAMP-1 on their surface, which is responsible for the resilient adhesion to surrounding E-selectin (cell adhesion molecule) expressing cells. Studies of the knockdown of LAMP-3 have shown a decline in the invasion capacity of cancer cells.93 Investigation of the knockdown of the PERK/ATF-4/LAMP-3 pathway of the UPR has demonstrated the role of PERK arm in the radio resistance of breast malignancies.129 In extracellular matrix containing detached mammary epithelial cells, PERK has been shown to be pro-survival for malignant cells, as it induces the shield of autophagy.130 Thus, the PERK arm of the UPR in alliance with autophagy supports the survival attempt of tumor cells under stressful conditions.131, 132
Role of IRE-1 in breast malignancy
There are reports of increased XBP-1 expression and their unconventional splicing in breast cancer, which is a downstream effector of IRE-1.133 Lin et al.134 demonstrated that in stressed cells, the ability of the UPR to navigate the cells through pro-survival or pro-death was largely a mandate of IRE-1 activity. The stimulation of breast cancer cells by 17β-estradiol (E2) treatment specifically upregulates XBP-1.134, 135 In a recent report by Xi Chen et al.,136 the levels of XBP-1 expression were easily detected in several breast cancer cell lines with triple-negative breast cancer (TNBC; tumors not expressing genes for estrogen receptor, progesterone receptor and human epidermal growth factor receptor 2), where they form an axis in tumor development and progression. In cases of anti-estrogen-resistant breast malignancies, XBP-1 expression levels are high with co-expression of the estrogen receptor alpha (ER-α) protein, a nuclear receptor protein expressed in ~70% of breast cancers.137 Overexpression cases of XBP-1 (S) make cells resistant to tumor-associated macrophages and ER-α targeting receptor antagonist (anti-estrogen), fulvestrant.138 NF-κB is also co-expressed in higher numbers with XBP-1139 and its signaling is increased by XBP-1 in anti-estrogen-resistant breast cancer cells.137 These findings provide evidence that XBP-1 promotes anti-estrogen resistance in breast malignancies, which is dependent on NF-κB activation.
Role of ATF-6 in breast malignancy
Prolonged ATF-6 activation is related to other UPR transducers under the condition of extended stress to activate the transcription of CHOP, thereby acting as a pro-death factor. Research conducted by Okada et al.43 in HeLa cells suggests a connection between the ATF-6 and the PERK arms of the UPR by converging at the activation of CHOP. Michallet et al.140 showed using knockdown studies in myeloma cells that the targeted loss of ATF-6 was pro-death. The ATF-6 arm of the UPR, when expressed at low levels, plays anticipatory roles in cell destruction.141 In one study on quiescent disseminated cancer cells, ATF-6 is identified as a pivotal transcription factor for their resilience to chemotherapy. This pitfall is created by ATF-6 through the upregulation of Rheb and mTOR activation.142 We still lack scientific data on the role of ATF-6 in cancer.
Inflammatory responses in breast cancer
Numerous documents note the escalating activity of NF-κB in breast malignancies.143, 144 Studies show that the activation of NF-κB might be one of the early events during breast malignancy development.145, 146 CD44, a unique cell surface glycoprotein, is upregulated in tumor cells.147, 148, 149 Smith et al.150, 151 have shown that the activity of a cis-acting element, conserved region 1, of CD44 is modulated by NF-κB.
Aging decreases the UPR while breast malignancy potentiates it: the UPR helps whom?
Breast cancer is a variegated malignancy displaying diversity within and between tumors, among patients and the age of the individual. There is an intricate alliance forged between age-related frailty and breast malignancy. There are many reports on cases in which the incidence of breast cancer rises exponentially with aging.152 Aging unequivocally mitigates the overall density of the breast and at the same time increases the cellular mass of adipose tissue,153 thereby causing a change in the microenvironment of the breast.154 The current review is an attempt to underline the molecular signatures inside the breast cancer cell that can help us to understand the relationship established between UPR and aging. However, there is a conundrum: aging-implicated ER stress is not decreased by the UPR; rather, the burdened cell is driven toward the treacherous path of apoptosis. We have discussed numerous cases in this review that support the UPR-driven activation of pro-death transcription factors engendered by aging. When we discuss the involvement of the UPR in the escalation of breast malignancy with the progression of aging, there is a sudden dearth of data. The UPR does not abrogate malignancy; rather, the cancer cells exploit the UPR transducers to become immortal by activating pro-survival signals. Therefore, it is plausible to discuss the crucial roles of aging and the UPR in unlocking the lesser-known aspects of breast malignancy.
The first conundrum worth mentioning here relates to the status of GRP78. Aging leads to the diminished expression pattern of GRP78, whereas in breast malignancy there is a surge in its expression. GRP78 is not only localized with the ER transmembrane UPR transducers, but there is mounting evidence of a subfraction of GRP78 being localized on the cell surface of tumor cells.155 Cases support the protective role of GRP78 for human breast cancer cells, where GRP78 confers endocrine resistance to the breast tumor.156 Some of the mechanistic details of GRP78 as an agent to keep cancer cells fertile come from its involvement in inhibiting apoptosis by preventing the cleavage of pro-caspase-7, inhibition of pro-apoptotic BCL2-family proteins and decreasing ER stress by maintaining low levels of misfolded proteins in the ER lumen, which again marks the inhibition of a pro-apoptotic UPR.117, 157, 158 The overexpression of GRP78 in breast cancer can be measured in its prognosis. Additionally, GRP78 has been implicated in chemoresistance to anticancer treatments; therefore, the levels of this ER chaperone may serve as a novel biomarker for chemoresponsiveness in breast malignancy.159
The novel substrate for PERK, NRF2, has been designated the master regulator of cytoprotective genes.160, 161 Aging abates the interactions of NRF2 with its target sequence, antioxidant response element, thereby inhibiting the role of NRF2 in responding to stressors, which favors age-related frailty.162 In breast cancer, NRF2 supports tumorigenesis and chemoresistance by affecting several molecular targets in the cell.163, 164 A plausible explanation for this paradoxical behavior of NRF2 would be the selective activation of the PERK arm of UPR by tumor cells to sustain the limiting microenvironment of malignancy.
The UPR signal transducer, ATF-6, upregulates the mTOR pathway in cancer cells.142 Studies have proven that the mTOR pathway is fueled up during aging.165, 166 This pathway and its target, the phosphoinositol 3 kinase (PI3K)/Akt/mammalian pathway, have also been well documented to promote tumor proliferation in endocrine-resistant breast cancer.167 The mTOR pathway is therefore a key target of crosstalks between the UPR and aging that sustains breast malignancy. Hence, a reduction in aging-linked inflammation by affecting mTOR could be a potential mechanism for interfering with aging-related malignancy.165, 166 More evidence of molecular cross-linking involves NF-κB, which is a hallmark of inflammatory responses and is activated through all three arms of the UPR: PERK, IRE-1 and ATF-6. There are accumulating reports that support the unabated transcriptional activity of NF-κB in different tissues with aging.104, 168, 169, 170 The work of Smith et al.151 in triple-negative breast cancer cells clearly shows that the inhibition of NF-κB can repress the expression of CD44 thereby mitigating the cell proliferation and invasiveness of breast cancer cells.
The UPR and hexosamine biosynthetic pathway in aging and breast cancer: a promise against agony
Increasing evidence suggests an increase in life span coupled with a decrease in cancer pathology by a considerable percentage under calorie restriction (CR), that is, restricting the intake of calories without prompting malnutrition.171, 172, 173 The effect of CR is effectively sensed at the cellular level through a well-studied metabolic pathway, the hexosamine biosynthetic pathway (HBP).174
The HBP produces the metabolite nucleotide sugar uridine diphosphate-N-acetylglucosamine (UDP-GlcNAc), which acts as the substrate for the process of N-glycosylation and O-glycosylation (O-GlcNAc), a mandatory step in protein folding within the ER lumen.175 Tumor progression demands extra glucose and glutamine to meet the needs of the tumor under conditions of nutritional deprivation. As studied in cases of breast cancer, the resulting hyperglycosylation in stressful ER conditions imposes a demand for substrate requirements on the HBP, leading to an increase in HBP flux.176, 177, 178 The resulting UPR shows an alliance with the HBP, in which the transcription of the first and important rate-limiting enzyme of the HBP, glutamine:fructose-6-phosphate aminotransferase 1 (GFAT-1), is directly stimulated by XBP-1. XBP-1 acts as an upstream activator of several genes encoding important enzymes in the HBP, thereby leading to a rise in O-GlcNAc protein modification during ER stress.179 In a separate study in chemoresistant basal-like/triple-negative breast cancer, higher levels of O-GlcNAc protein modification have been observed.180 Chaveroux et al.181 showed in their study that the PERK/eIF2α/ATF-4 arm of the UPR also regulates HBP. They demonstrated the involvement of ATF-4 in controlling the abundance of GFAT-1, which further evokes protein modification under conditions of nutritional stress.182 Together, these findings strongly corroborate the cytoprotective aspect of the UPR–HBP alliance. RNA interference studies in breast cancer cells report that the downregulation of GFAT-1 has anti-progression and anti-invasion effects.176
Aging induces rise in nutrient sensing, leading to increased HBP flux which is reasoned for the onset of insulin resistance and age-related maladies.183 The exhausted ER protein homeostasis during aging can be ameliorated by modulating GFAT-1, as shown in a study on C. elegans, where a gain-of-function mutation in GFAT-1-induced ERAD and autophagy, resulting in better health and longevity.184
The role of CR in impeding aging-associated pathologies has been demonstrated in young calorie-restricted rats, where insulin sensitivity escalated with decreasing hexosamine levels, proving the participation of the HBP in features of aging.185 A recent report by Busti et al.186 advocates CR in relieving ER stress. Thus, CR can be translated into a strategy, which is an effective and reproducible intervention against the alarmingly increasing cases of aging-driven cancers.
Conclusion
Until now, the cause of increasing risk of breast cancer in women over 50 years of age was a conundrum. Some explanations have come through studies that support the secretions from aging stromal cells supporting pre-cancer cells; also, aging potentiates pro-inflammatory pathways that provide selective advantages to cancer cells.187, 188, 189, 190 The data on the direct involvement of the UPR in aging-driven breast cancer cases are seemingly feeble. There is a need to study this aspect of the story. Aging-linked upregulation of the UPR drives cells toward pro-death. Breast malignancy-linked activation of the UPR mitigates the pro-death attempt and supports the immortality of cancer cells.
There are numerous reports that suggest that therapeutic interventions of UPR transducers and their effectors can diminish the supportive effect on tumors.191 We need a more systemic approach that is not reductionist, but at the same time, we should focus on various aspects of aging, breast malignancy and the molecular mechanism of the UPR so that human intervention can utilize new findings in breast cancer.
References
American Cancer Society. Breast Cancer Facts & Figures 2015-2016. American Cancer Society, Inc: Atlanta, 2015.
Wei Q, Matanoski GM, Farmer ER, Hedayati MA, Grossman L . DNA repair and aging in basal cell carcinoma: a molecular epidemiology study. Proc Natl Acad Sci USA 1993; 90: 1614–1618.
Niedermuller H . Age dependency of DNA repair in rats after DNA damage by carcinogens. Mech Ageing Dev 1982; 19: 259–271.
Gu J, Spitz MR, Zhao H, Lin J, Grossman HB, Dinney CP et al. Roles of tumor suppressor and telomere maintenance genes in cancer and aging—an epidemiological study. Carcinogenesis 2005; 26: 1741–1747.
Bastien RRL, Rodríguez-Lescure Á, Ebbert MTW, Prat A, Munárriz B, Rowe L et al. PAM50 breast cancer subtyping by RT-qPCR and concordance with standard clinical molecular markers. BMC Med Genomics 2012; 5: 44–44.
Yau C, Fedele V, Roydasgupta R, Fridlyand J, Hubbard A, Gray JW et al. Aging impacts transcriptomes but not genomes of hormone-dependent breast cancers. Breast Cancer Res 2007; 9: R59.
The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012; 490: 61–70.
Garbe JC, Pepin F, Pelissier FA, Sputova K, Fridriksdottir AJ, Guo DE et al. Accumulation of multipotent progenitors with a basal differentiation bias during aging of human mammary epithelia. Cancer Res 2012; 72: 3687–3701.
Haakensen VD, Lingjaerde OC, Luders T, Riis M, Prat A, Troester MA et al. Gene expression profiles of breast biopsies from healthy women identify a group with claudin-low features. BMC Med Genomics 2011; 4: 77.
Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P . Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 2000; 101: 249–258.
Ellgaard L, Helenius A . Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol 2003; 4: 181–191.
Munro S, Pelham HR . An Hsp70-like protein in the ER: identity with the 78 kd glucose-regulated protein and immunoglobulin heavy chain binding protein. Cell 1986; 46: 291–300.
Naidoo N . ER and aging-protein folding and the ER stress response. Ageing Res Rev 2009; 8: 150–159.
Erickson RR, Dunning LM, Holtzman JL . The effect of aging on the chaperone concentrations in the hepatic, endoplasmic reticulum of male rats: the possible role of protein misfolding due to the loss of chaperones in the decline in physiological function seen with age. J Gerontol A Biol Sci Med Sci 2006; 61: 435–443.
Schroder M, Kaufman RJ . The mammalian unfolded protein response. Annu Rev Biochem 2005; 74: 739–789.
Sundfør K, Lyng H, Rofstad EK . Tumour hypoxia and vascular density as predictors of metastasis in squamous cell carcinoma of the uterine cervix. Br J Cancer 1998; 78: 822–827.
Fels DR, Koumenis C . The PERK/eIF2alpha/ATF4 module of the UPR in hypoxia resistance and tumor growth. Cancer Biol Ther 2006; 5: 723–728.
Wang Y, Alam GN, Ning Y, Visioli F, Dong Z, Nör JE et al. The unfolded protein response induces the angiogenic switch in human tumor cells through the PERK/ATF4 pathway. Cancer Res 2012; 72: 5396–5406.
Morris JA, Dorner AJ, Edwards CA, Hendershot LM, Kaufman RJ . Immunoglobulin binding protein (BiP) function is required to protect cells from endoplasmic reticulum stress but is not required for the secretion of selective proteins. J Biol Chem 1997; 272: 4327–4334.
McCracken AA, Brodsky JL . Assembly of ER-associated protein degradation in vitro: dependence on cytosol, calnexin, and ATP. J Cell Biol 1996; 132: 291–298.
Haynes CM, Caldwell S, Cooper AA . An HRD/DER-independent ER quality control mechanism involves Rsp5p-dependent ubiquitination and ER-Golgi transport. J Cell Biol 2002; 158: 91–101.
Yorimitsu T, Nair U, Yang Z, Klionsky DJ . Endoplasmic reticulum stress triggers autophagy. J Biol Chem 2006; 281: 30299–30304.
Harding HP, Zhang Y, Ron D . Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999; 397: 271–274.
Dever TE . Gene-specific regulation by general translation factors. Cell 2002; 108: 545–556.
Hinnebusch AG . 5 Mechanism and regulation of initiator methionyl-tRNA binding to ribosomes. In: Sonenberg N, Hershey JWB, Mathews MB (eds). Translational Control of Gene Expression. CSHL Press: Cold Spring Harbor, NY, USA, 2000, pp 185–243.
Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 2000; 6: 1099–1108.
Baumeister P, Luo S, Skarnes WC, Sui G, Seto E, Shi Y et al. Endoplasmic reticulum stress induction of the Grp78/BiP promoter: activating mechanisms mediated by YY1 and its interactive chromatin modifiers. Mol Cell Biol 2005; 25: 4529–4540.
Vattem KM, Wek RC . Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci USA 2004; 101: 11269–11274.
Lu PD, Harding HP, Ron D . Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol 2004; 167: 27–33.
Ron D . Translational control in the endoplasmic reticulum stress response. J Clin Invest 2002; 110: 1383–1388.
Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 2003; 11: 619–633.
Palam LR, Baird TD, Wek RC . Phosphorylation of eIF2 facilitates ribosomal bypass of an inhibitory upstream ORF to enhance CHOP translation. J Biol Chem 2011; 286: 10939–10949.
Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev 2004; 18: 3066–3077.
Song B, Scheuner D, Ron D, Pennathur S, Kaufman RJ . Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J Clin Invest 2008; 118: 3378–3389.
Yoshida H, Matsui T, Hosokawa N, Kaufman RJ, Nagata K, Mori K . A time-dependent phase shift in the mammalian unfolded protein response. Dev Cell 2003; 4: 265–271.
Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K . XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001; 107: 881–891.
Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 2004; 21: 81–93.
Cross BC, Bond PJ, Sadowski PG, Jha BK, Zak J, Goodman JM et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc Natl Acad Sci USA 2012; 109: E869–E878.
Kimata Y, Ishiwata-Kimata Y, Ito T, Hirata A, Suzuki T, Oikawa D et al. Two regulatory steps of ER-stress sensor Ire1 involving its cluster formation and interaction with unfolded proteins. J Cell Biol 2007; 179: 75–86.
Gardner BM, Walter P . Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science 2011; 333: 1891–1894.
Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell 2000; 6: 1355–1364.
Yoshida H, Haze K, Yanagi H, Yura T, Mori K . Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J Biol Chem 1998; 273: 33741–33749.
Okada T, Yoshida H, Akazawa R, Negishi M, Mori K . Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem J 2002; 366: 585–594.
Lee AH, Iwakoshi NN, Glimcher LH . XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 2003; 23: 7448–7459.
Wu J, Rutkowski DT, Dubois M, Swathirajan J, Saunders T, Wang J et al. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev Cell 2007; 13: 351–364.
Ikeyama S, Wang XT, Li J, Podlutsky A, Martindale JL, Kokkonen G et al. Expression of the pro-apoptotic gene gadd153/chop is elevated in liver with aging and sensitizes cells to oxidant injury. J Biol Chem 2003; 278: 16726–16731.
Elmore S . Apoptosis: a review of programmed cell death. Toxicol Pathol 2007; 35: 495–516.
Nakagawa T, Yuan J . Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J Cell Biol 2000; 150: 887–894.
Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 2000; 403: 98–103.
Rao RV, Hermel E, Castro-Obregon S, del Rio G, Ellerby LM, Ellerby HM et al. Coupling endoplasmic reticulum stress to the cell death program. Mechanism of caspase activation. J Biol Chem 2001; 276: 33869–33874.
Fawcett TW, Martindale JL, Guyton KZ, Hai T, Holbrook NJ . Complexes containing activating transcription factor (ATF)/cAMP-responsive-element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response. Biochem J 1999; 339 (Pt 1): 135–141.
Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H . TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J 2005; 24: 1243–1255.
Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev 1998; 12: 982–995.
Zou H, Henzel WJ, Liu X, Lutschg A, Wang X . Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 1997; 90: 405–413.
Pan G, O’Rourke K, Dixit VM . Caspase-9, Bcl-XL, and Apaf-1 form a ternary complex. J Biol Chem 1998; 273: 5841–5845.
Wang XZ, Ron D . Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP kinase. Science 1996; 272: 1347–1349.
Granger GA, Shacks SJ, Williams TW, Kolb WP . Lymphocyte in vitro cytotoxicity: specific release of lymphotoxin-like materials from tuberculin-sensitive lymphoid cells. Nature 1969; 221: 1155–1157.
Suda T, Takahashi T, Golstein P, Nagata S . Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family. Cell 1993; 75: 1169–1178.
Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000; 287: 664–666.
Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K et al. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev 2002; 16: 1345–1355.
Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V et al. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 2008; 453: 807–811.
Cullinan SB, Diehl JA . PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J Biol Chem 2004; 279: 20108–20117.
Deniaud A, Sharaf el dein O, Maillier E, Poncet D, Kroemer G, Lemaire C et al. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 2008; 27: 285–299.
Deng J, Lu PD, Zhang Y, Scheuner D, Kaufman RJ, Sonenberg N et al. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol Cell Biol 2004; 24: 10161–10168.
Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH . Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol Cell Biol 2006; 26: 3071–3084.
Zheng GF, Cai Z, Meng XK, Zhang Y, Zhu W, Pang XY et al. Unfolded protein response mediated JNK/AP-1 signal transduction, a target for ovarian cancer treatment. Int J Clin Exp Pathol 2015; 8: 6505–6511.
Yamazaki H, Hiramatsu N, Hayakawa K, Tagawa Y, Okamura M, Ogata R et al. Activation of the Akt-NF-kappaB pathway by subtilase cytotoxin through the ATF6 branch of the unfolded protein response. J Immunol 2009; 183: 1480–1487.
Hara K, Yonezawa K, Kozlowski MT, Sugimoto T, Andrabi K, Weng QP et al. Regulation of eIF-4E BP1 phosphorylation by mTOR. J Biol Chem 1997; 272: 26457–26463.
Hannan KM, Brandenburger Y, Jenkins A, Sharkey K, Cavanaugh A, Rothblum L et al. mTOR-dependent regulation of ribosomal gene transcription requires S6K1 and is mediated by phosphorylation of the carboxy-terminal activation domain of the nucleolar transcription factor UBF. Mol Cell Biol 2003; 23: 8862–8877.
Helliwell SB, Howald I, Barbet N, Hall MN . TOR2 is part of two related signaling pathways coordinating cell growth in Saccharomyces cerevisiae. Genetics 1998; 148: 99–112.
Soukas AA, Kane EA, Carr CE, Melo JA, Ruvkun G . Rictor/TORC2 regulates fat metabolism, feeding, growth, and life span in Caenorhabditis elegans. Genes Dev 2009; 23: 496–511.
Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010; 465: 942–946.
Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P . mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 2007; 450: 736–740.
Kato H, Nakajima S, Saito Y, Takahashi S, Katoh R, Kitamura M . mTORC1 serves ER stress-triggered apoptosis via selective activation of the IRE1–JNK pathway. Cell Death Differ 2012; 19: 310–320.
Lee DF, Kuo HP, Chen CT, Hsu JM, Chou CK, Wei Y et al. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 2007; 130: 440–455.
Klionsky DJ, Cregg JM, Dunn WA Jr, Emr SD, Sakai Y, Sandoval IV et al. A unified nomenclature for yeast autophagy-related genes. Dev Cell 2003; 5: 539–545.
Salazar M, Carracedo A, Salanueva ÍJ, Hernández-Tiedra S, Lorente M, Egia A et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J Clin Invest 2009; 119: 1359–1372.
Chandrika BB, Yang C, Ou Y, Feng X, Muhoza D, Holmes AF et al. Endoplasmic reticulum stress-induced autophagy provides cytoprotection from chemical hypoxia and oxidant injury and ameliorates renal ischemia-reperfusion injury. PLoS ONE 2015; 10: e0140025.
Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169: 425–434.
Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441: 880–884.
Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003; 112: 1809–1820.
Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H et al. ER stress (PERK//eIF2[alpha] phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 2006; 14: 230–239.
Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E . Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 2005; 1: 84–91.
Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P . Distinct classes of phosphatidylinositol 3'-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem 2000; 275: 992–998.
Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19: 5720–5728.
Chen JW, Murphy TL, Willingham MC, Pastan I, August JT . Identification of two lysosomal membrane glycoproteins. J Cell Biol 1985; 101: 85–95.
Andrejewski N, Punnonen EL, Guhde G, Tanaka Y, Lullmann-Rauch R, Hartmann D et al. Normal lysosomal morphology and function in LAMP-1-deficient mice. J Biol Chem 1999; 274: 12692–12701.
Lee E-J, Park K-S, Jeon I-S, Choi J-W, Lee S-J, Choy HE et al. LAMP-3 (lysosome-associated membrane protein 3) promotes the intracellular proliferation of Salmonella typhimurium. Mol Cells 2016; 39: 566–572.
de Saint-Vis B, Vincent J, Vandenabeele S, Vanbervliet B, Pin JJ, Ait-Yahia S et al. A novel lysosome-associated membrane glycoprotein, DC-LAMP, induced upon DC maturation, is transiently expressed in MHC class II compartment. Immunity 1998; 9: 325–336.
Rzymski T, Milani M, Pike L, Buffa F, Mellor HR, Winchester L et al. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene 2010; 29: 4424–4435.
Saha T . LAMP2A overexpression in breast tumors promotes cancer cell survival via chaperone-mediated autophagy. Autophagy 2012; 8: 1643–1656.
Liao X, Chen Y, Liu D, Li F, Li X, Jia W . High expression of LAMP3 is a novel biomarker of poor prognosis in patients with esophageal squamous cell carcinoma. Int J Mol Sci 2015; 16: 17655–17667.
Nagelkerke A, Bussink J, Mujcic H, Wouters BG, Lehmann S, Sweep FC et al. Hypoxia stimulates migration of breast cancer cells via the PERK/ATF4/LAMP3-arm of the unfolded protein response. Breast Cancer Res 2013; 15: R2.
Mujcic H, Rzymski T, Rouschop KMA, Koritzinsky M, Milani M, Harris AL et al. Hypoxic activation of the unfolded protein response (UPR) induces expression of the metastasis-associated gene LAMP3. Radiother Oncol 2009; 92: 450–459.
Margariti A, Li H, Chen T, Martin D, Vizcay-Barrena G, Alam S et al. XBP1 mRNA splicing triggers an autophagic response in endothelial cells through BECLIN-1 transcriptional activation. J Biol Chem 2013; 288: 859–872.
Tang SW, Chen CY, Klase Z, Zane L, Jeang KT . The cellular autophagy pathway modulates human T-cell leukemia virus type 1 replication. J Virol 2013; 87: 1699–1707.
Djavaheri-Mergny M, Amelotti M, Mathieu J, Besancon F, Bauvy C, Codogno P . Regulation of autophagy by NFkappaB transcription factor and reactives oxygen species. Autophagy 2007; 3: 390–392.
Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 2009; 20: 1981–1991.
Hussain SG, Ramaiah KV . Reduced eIF2alpha phosphorylation and increased proapoptotic proteins in aging. Biochem Biophys Res Commun 2007; 355: 365–370.
Naidoo N, Ferber M, Master M, Zhu Y, Pack AI . Aging impairs the unfolded protein response to sleep deprivation and leads to proapoptotic signaling. J Neurosci 2008; 28: 6539–6548.
Paz Gavilan M, Vela J, Castano A, Ramos B, del Rio JC, Vitorica J et al. Cellular environment facilitates protein accumulation in aged rat hippocampus. Neurobiol Aging 2006; 27: 973–982.
Nuss JE, Choksi KB, DeFord JH, Papaconstantinou J . Decreased enzyme activities of chaperones PDI and BiP in aged mouse livers. Biochem Biophys Res Commun 2008; 365: 355–361.
Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997; 275: 90–94.
Adler AS, Sinha S, Kawahara TL, Zhang JY, Segal E, Chang HY . Motif module map reveals enforcement of aging by continual NF-kappaB activity. Genes Dev 2007; 21: 3244–3257.
Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S . Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol 2004; 14: 885–890.
Syntichaki P, Troulinaki K, Tavernarakis N . eIF4E function in somatic cells modulates ageing in Caenorhabditis elegans. Nature 2007; 445: 922–926.
Brunk UT, Terman A . Lipofuscin: mechanisms of age-related accumulation and influence on cell function. Free Radic Biol Med 2002; 33: 611–619.
Terman A, Dalen H, Brunk UT . Ceroid/lipofuscin-loaded human fibroblasts show decreased survival time and diminished autophagocytosis during amino acid starvation. Exp Gerontol 1999; 34: 943–957.
Carames B, Taniguchi N, Otsuki S, Blanco FJ, Lotz M . Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum 2010; 62: 791–801.
Holtz WA, O'Malley KL . Parkinsonian mimetics induce aspects of unfolded protein response in death of dopaminergic neurons. J Biol Chem 2003; 278: 19367–19377.
Hoozemans JJ, Veerhuis R, Van Haastert ES, Rozemuller JM, Baas F, Eikelenboom P et al. The unfolded protein response is activated in Alzheimer's disease. Acta Neuropathol 2005; 110: 165–172.
Mandriota SJ, Pepper MS . Regulation of angiopoietin-2 mRNA levels in bovine microvascular endothelial cells by cytokines and hypoxia. Circ Res 1998; 83: 852–859.
Liu Y, Cox SR, Morita T, Kourembanas S . Hypoxia regulates vascular endothelial growth factor gene expression in endothelial cells. Identification of a 5' enhancer. Circ Res 1995; 77: 638–643.
Das B, Yeger H, Tsuchida R, Torkin R, Gee MF, Thorner PS et al. A hypoxia-driven vascular endothelial growth factor/Flt1 autocrine loop interacts with hypoxia-inducible factor-1alpha through mitogen-activated protein kinase/extracellular signal-regulated kinase 1/2 pathway in neuroblastoma. Cancer Res 2005; 65: 7267–7275.
Wang Y, Ning Y, Alam GN, Jankowski BM, Dong Z, Nör JE et al. Amino acid deprivation promotes tumor angiogenesis through the GCN2/ATF4 pathway. Neoplasia 2013; 15: 989–997.
Yao X, Liu H, Zhang X, Zhang L, Li X, Wang C et al. Cell surface GRP78 accelerated breast cancer cell proliferation and migration by activating STAT3. PLoS ONE 2015; 10: e0125634.
Fu Y, Li J, Lee AS . GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res 2007; 67: 3734–3740.
Bobrovnikova-Marjon E, Grigoriadou C, Pytel D, Zhang F, Ye J, Koumenis C et al. PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene 2010; 29: 3881–3895.
Bi M, Naczki C, Koritzinsky M, Fels D, Blais J, Hu N et al. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J 2005; 24: 3470–3481.
Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA . Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol 2003; 23: 7198–7209.
Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 1999; 13: 76–86.
Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 1997; 236: 313–322.
Chan K, Han XD, Kan YW . An important function of Nrf2 in combating oxidative stress: detoxification of acetaminophen. Proc Natl Acad Sci USA 2001; 98: 4611–4616.
Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D . Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell 2000; 5: 897–904.
Wild AC, Moinova HR, Mulcahy RT . Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J Biol Chem 1999; 274: 33627–33636.
Iida K, Itoh K, Kumagai Y, Oyasu R, Hattori K, Kawai K et al. Nrf2 is essential for the chemopreventive efficacy of oltipraz against urinary bladder carcinogenesis. Cancer Res 2004; 64: 6424–6431.
Syu JP, Chi JT, Kung HN . Nrf2 is the key to chemotherapy resistance in MCF7 breast cancer cells under hypoxia. Oncotarget 2016; 7: 14659–14672.
Sawada R, Lowe JB, Fukuda M . E-selectin-dependent adhesion efficiency of colonic carcinoma cells is increased by genetic manipulation of their cell surface lysosomal membrane glycoprotein-1 expression levels. J Biol Chem 1993; 268: 12675–12681.
Nagelkerke A, Bussink J, van der Kogel AJ, Sweep FC, Span PN . The PERK/ATF4/LAMP3-arm of the unfolded protein response affects radioresistance by interfering with the DNA damage response. Radiother Oncol 2013; 108: 415–421.
Avivar-Valderas A, Salas E, Bobrovnikova-Marjon E, Diehl JA, Nagi C, Debnath J et al. PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol Cell Biol 2011; 31: 3616–3629.
Ding WX, Ni HM, Gao W, Hou YF, Melan MA, Chen X et al. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem 2007; 282: 4702–4710.
Ye J, Kumanova M, Hart LS, Sloane K, Zhang H, De Panis DN et al. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J 2010; 29: 2082–2096.
Hong SY, Hagen T . Multiple myeloma Leu167Ile (c.499C>A) mutation prevents XBP1 mRNA splicing. Br J Haematol 2013; 161: 898–901.
Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B et al. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007; 318: 944–949.
Wang DY, Fulthorpe R, Liss SN, Edwards EA . Identification of estrogen-responsive genes by complementary deoxyribonucleic acid microarray and characterization of a novel early estrogen-induced gene: EEIG1. Mol Endocrinol 2004; 18: 402–411.
Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature 2014; 508: 103–107.
Hu R, Warri A, Jin L, Zwart A, Riggins RB, Fang H-B et al. NF-κB signaling is required for XBP1 (unspliced and spliced)-mediated effects on antiestrogen responsiveness and cell fate decisions in breast cancer. Mol Cell Biol 2015; 35: 379–390.
Gomez BP, Riggins RB, Shajahan AN, Klimach U, Wang A, Crawford AC et al. Human X-box binding protein-1 confers both estrogen independence and antiestrogen resistance in breast cancer cell lines. FASEB J 2007; 21: 4013–4027.
Zhu Y, Singh B, Hewitt S, Liu A, Gomez B, Wang A et al. Expression patterns among interferon regulatory factor-1, human X-box binding protein-1, nuclear factor kappa B, nucleophosmin, estrogen receptor-alpha and progesterone receptor proteins in breast cancer tissue microarrays. Int J Oncol 2006; 28: 67–76.
Michallet AS, Mondiere P, Taillardet M, Leverrier Y, Genestier L, Defrance T . Compromising the unfolded protein response induces autophagy-mediated cell death in multiple myeloma cells. PLoS ONE 2011; 6: e25820.
Morishima N, Nakanishi K, Nakano A . Activating transcription factor-6 (ATF6) mediates apoptosis with reduction of myeloid cell leukemia sequence 1 (Mcl-1) protein via induction of WW domain binding protein 1. J Biol Chem 2011; 286: 35227–35235.
Schewe DM, Aguirre-Ghiso JA . ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc Natl Acad Sci USA 2008; 105: 10519–10524.
Cogswell PC, Guttridge DC, Funkhouser WK, Baldwin AS Jr . Selective activation of NF-kappa B subunits in human breast cancer: potential roles for NF-kappa B2/p52 and for Bcl-3. Oncogene 2000; 19: 1123–1131.
Nakshatri H, Bhat-Nakshatri P, Martin DA, Goulet RJ Jr, Sledge GW Jr . Constitutive activation of NF-kappaB during progression of breast cancer to hormone-independent growth. Mol Cell Biol 1997; 17: 3629–3639.
Dejardin E, Bonizzi G, Bellahcene A, Castronovo V, Merville MP, Bours V . Highly-expressed p100/p52 (NFKB2) sequesters other NF-kappa B-related proteins in the cytoplasm of human breast cancer cells. Oncogene 1995; 11: 1835–1841.
Kim DW, Sovak MA, Zanieski G, Nonet G, Romieu-Mourez R, Lau AW et al. Activation of NF-kappaB/Rel occurs early during neoplastic transformation of mammary cells. Carcinogenesis 2000; 21: 871–879.
Bourguignon LY, Peyrollier K, Xia W, Gilad E . Hyaluronan-CD44 interaction activates stem cell marker Nanog, Stat-3-mediated MDR1 gene expression, and ankyrin-regulated multidrug efflux in breast and ovarian tumor cells. J Biol Chem 2008; 283: 17635–17651.
Hiraga T, Ito S, Nakamura H . Cancer stem-like cell marker CD44 promotes bone metastases by enhancing tumorigenicity, cell motility, and hyaluronan production. Cancer Res 2013; 73: 4112–4122.
Honeth G, Bendahl PO, Ringner M, Saal LH, Gruvberger-Saal SK, Lovgren K et al. The CD44+/CD24- phenotype is enriched in basal-like breast tumors. Breast Cancer Res 2008; 10: R53.
Smith SM, Cai L . Cell specific CD44 expression in breast cancer requires the interaction of AP-1 and NFkappaB with a novel cis-element. PLoS ONE 2012; 7: e50867.
Smith SM, Lyu YL, Cai L . NF-kappaB affects proliferation and invasiveness of breast cancer cells by regulating CD44 expression. PLoS ONE 2014; 9: e106966.
Milanese TR, Hartmann LC, Sellers TA, Frost MH, Vierkant RA, Maloney SD et al. Age-related lobular involution and risk of breast cancer. J Natl Cancer Inst 2006; 98: 1600–1607.
McCormack VA, Perry NM, Vinnicombe SJ, Dos Santos Silva I . Changes and tracking of mammographic density in relation to Pike's model of breast tissue aging: a UK longitudinal study. Int J Cancer 2010; 127: 452–461.
LaBarge MA, Nelson CM, Villadsen R, Fridriksdottir A, Ruth JR, Stampfer MR et al. Human mammary progenitor cell fate decisions are products of interactions with combinatorial microenvironments. Integr Biol 2009; 1: 70–79.
Zhang Y, Liu R, Ni M, Gill P, Lee AS . Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. J Biol Chem 2010; 285: 15065–15075.
Zhou H, Zhang Y, Fu Y, Chan L, Lee AS . Novel mechanism of anti-apoptotic function of 78-kDa glucose-regulated protein (GRP78): endocrine resistance factor in breast cancer, through release of B-cell lymphoma 2 (BCL-2) from BCL-2-interacting killer (BIK). J Biol Chem 2011; 286: 25687–25696.
Reddy RK, Mao C, Baumeister P, Austin RC, Kaufman RJ, Lee AS . Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase-7 activation. J Biol Chem 2003; 278: 20915–20924.
Cook KL, Shajahan AN, Warri A, Jin L, Hilakivi-Clarke LA, Clarke R . Glucose-regulated protein 78 controls cross-talk between apoptosis and autophagy to determine antiestrogen responsiveness. Cancer Res 2012; 72: 3337–3349.
Lee E, Nichols P, Spicer D, Groshen S, Yu MC, Lee AS . GRP78 as a novel predictor of responsiveness to chemotherapy in breast cancer. Cancer Res 2006; 66: 7849–7853.
Lee JM, Calkins MJ, Chan K, Kan YW, Johnson JA . Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J Biol Chem 2003; 278: 12029–12038.
Liu XM, Peyton KJ, Ensenat D, Wang H, Hannink M, Alam J et al. Nitric oxide stimulates heme oxygenase-1 gene transcription via the Nrf2/ARE complex to promote vascular smooth muscle cell survival. Cardiovasc Res 2007; 75: 381–389.
Gounder SS, Kannan S, Devadoss D, Miller CJ, Whitehead KJ, Odelberg SJ et al. Impaired transcriptional activity of Nrf2 in age-related myocardial oxidative stress is reversible by moderate exercise training. PLoS ONE 2012; 7: e45697.
Ikeda H, Nishi S, Sakai M . Transcription factor Nrf2/MafK regulates rat placental glutathione S-transferase gene during hepatocarcinogenesis. Biochem J 2004; 380: 515–521.
Nioi P, Nguyen T . A mutation of Keap1 found in breast cancer impairs its ability to repress Nrf2 activity. Biochem Biophys Res Commun 2007; 362: 816–821.
Powers RW 3rd, Kaeberlein M, Caldwell SD, Kennedy BK, Fields S . Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev 2006; 20: 174–184.
Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009; 460: 392–395.
Klos KS, Wyszomierski SL, Sun M, Tan M, Zhou X, Li P et al. ErbB2 increases vascular endothelial growth factor protein synthesis via activation of mammalian target of rapamycin/p70S6K leading to increased angiogenesis and spontaneous metastasis of human breast cancer cells. Cancer Res 2006; 66: 2028–2037.
Xiao ZQ, Majumdar AP . Induction of transcriptional activity of AP-1 and NF-kappaB in the gastric mucosa during aging. Am J Physiol Gastrointest Liver Physiol 2000; 278: G855–G865.
Helenius M, Hanninen M, Lehtinen SK, Salminen A . Aging-induced up-regulation of nuclear binding activities of oxidative stress responsive NF-kB transcription factor in mouse cardiac muscle. J Mol Cell Cardiol 1996; 28: 487–498.
Korhonen P, Helenius M, Salminen A . Age-related changes in the regulation of transcription factor NF-κB in rat brain. Neurosci Lett 1997; 225: 61–64.
Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009; 325: 201–204.
Harper JM, Leathers CW, Austad SN . Does caloric restriction extend life in wild mice? Aging Cell 2006; 5: 441–449.
Pugh TD, Oberley TD, Weindruch R . Dietary intervention at middle age: caloric restriction but not dehydroepiandrosterone sulfate increases lifespan and lifetime cancer incidence in mice. Cancer Res 1999; 59: 1642–1648.
Obici S, Wang J, Chowdury R, Feng Z, Siddhanta U, Morgan K et al. Identification of a biochemical link between energy intake and energy expenditure. J Clin Invest 2002; 109: 1599–1605.
Wells L, Vosseller K, Hart GW . Glycosylation of nucleocytoplasmic proteins: signal transduction and O-GlcNAc. Science 2001; 291: 2376–2378.
Caldwell SA, Jackson SR, Shahriari KS, Lynch TP, Sethi G, Walker S et al. Nutrient sensor O-GlcNAc transferase regulates breast cancer tumorigenesis through targeting of the oncogenic transcription factor FoxM1. Oncogene 2010; 29: 2831–2842.
Gu Y, Mi W, Ge Y, Liu H, Fan Q, Han C et al. GlcNAcylation plays an essential role in breast cancer metastasis. Cancer Res 2010; 70: 6344–6351.
Hazari YM, Bashir A, Haq EU, Fazili KM . Emerging tale of UPR and cancer: an essentiality for malignancy. Tumour Biol 2016; 37: 14381–14390.
Wang ZV, Deng Y, Gao N, Pedrozo Z, Li DL, Morales CR et al. Spliced X-box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway. Cell 2014; 156: 1179–1192.
Ferrer CM, Lynch TP, Sodi VL, Falcone JN, Schwab LP, Peacock DL et al. O-GlcNAcylation regulates cancer metabolism and survival stress signaling via regulation of HIF-1 pathway. Mol Cell 2014; 54: 820–831.
Chaveroux C, Sarcinelli C, Barbet V, Belfeki S, Barthelaix A, Ferraro-Peyret C et al. Nutrient shortage triggers the hexosamine biosynthetic pathway via the GCN2-ATF4 signalling pathway. Sci Rep 2016; 6: 27278.
Du Toit A . Post-translational modification: Sweetening protein quality control. Nat Rev Mol Cell Biol 2014; 15: 295–295.
Einstein FH, Fishman S, Bauman J, Thompson RF, Huffman DM, Atzmon G et al. Enhanced activation of a ‘nutrient-sensing’ pathway with age contributes to insulin resistance. FASEB J 2008; 22: 3450–3457.
Denzel MS, Storm J, Gutschmidt A, Baddi R, Hinze Y, Jarosch E et al. Hexosamine pathway metabolites enhance protein quality control and prolong life. Cell 156: 1167–1178.
Gazdag AC, Wetter TJ, Davidson RT, Robinson KA, Buse MG, Yee AJ et al. Lower calorie intake enhances muscle insulin action and reduces hexosamine levels. Am J Physiol Regul Integr Comp Physiol 2000; 278: R504–R512.
Busti S, Mapelli V, Tripodi F, Sanvito R, Magni F, Coccetti P et al. Respiratory metabolism and calorie restriction relieve persistent endoplasmic reticulum stress induced by calcium shortage in yeast. Sci Rep 2016; 6: 27942.
Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008; 133: 1019–1031.
Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008; 133: 1006–1018.
Ahmad A, Banerjee S, Wang Z, Kong D, Majumdar APN, Sarkar FH . Aging and inflammation: etiological culprits of cancer. Curr Aging Sci 2009; 2: 174–186.
Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004; 118: 285–296.
Zhou Y, Shu F, Liang X, Chang H, Shi L, Peng X et al. Ampelopsin induces cell growth inhibition and apoptosis in breast cancer cells through ROS generation and endoplasmic reticulum stress pathway. PLoS ONE 2014; 9: e89021.
Acknowledgements
We wholeheartedly thank all the authors whose research work has made this review possible. The research was supported by Yeungnam University research grants for the year 2016.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Minakshi, R., Rahman, S., Jan, A. et al. Implications of aging and the endoplasmic reticulum unfolded protein response on the molecular modality of breast cancer. Exp Mol Med 49, e389 (2017). https://doi.org/10.1038/emm.2017.215
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2017.215
This article is cited by
-
CD133 expression is associated with less DNA repair, better response to chemotherapy and survival in ER-positive/HER2-negative breast cancer
Breast Cancer Research and Treatment (2024)
-
mTOR inhibition abrogates human mammary stem cells and early breast cancer progression markers
Breast Cancer Research (2023)
-
Aging microenvironment and antitumor immunity for geriatric oncology: the landscape and future implications
Journal of Hematology & Oncology (2023)
