
Abstract
Genome-wide association studies have identified various genetic variants associated with complex disorders. However, these studies have commonly been conducted in a cross-sectional manner. Therefore, we performed a longitudinal exome-wide association study (EWAS) in a Japanese cohort. We aimed to identify genetic variants that confer susceptibility to hypertension using ~244 000 single-nucleotide variants (SNVs) and physiological data from 6026 Japanese individuals who underwent annual health check-ups for several years. After quality control, the association of hypertension with SNVs was tested using a generalized estimating equation model. Finally, our longitudinal EWAS detected seven hypertension-related SNVs that passed strict criteria. Among these variants, six SNVs were densely located at 12q24.1, and an East Asian-specific motif (haplotype) ‘CAAAA’ comprising five derived alleles was identified. Statistical analyses showed that the prevalence of hypertension in individuals with the East Asian-specific haplotype was significantly lower than that in individuals with the common haplotype ‘TGGGT’. Furthermore, individuals with the East Asian haplotype may be less susceptible to the adverse effects of smoking on hypertension. The longitudinal EWAS for the recessive model showed that a novel SNV, rs11917356 of COL6A5, was significantly associated with systolic blood pressure, and the derived allele at the SNV may have spread throughout East Asia in recent evolutionary time.
Similar content being viewed by others


Introduction
Hypertension is a major complex disorder affected by both genetic background and environmental factors, including lifestyle. In 2014, the Ministry of Health, Labour and Welfare reported that the estimated number of Japanese patients receiving medical treatment for hypertension had increased by ~10% since 2011 and was more than 10 million1 (http://www.mhlw.go.jp/english/database/db-hss/). Therefore, the exploration of genetic variants that confer susceptibility to hypertension is important for the personalized prevention of hypertension.
In recent decades, genome-wide association studies (GWASs) in various ethnic populations have identified genes, or loci, associated with hypertension.2, 3, 4, 5, 6, 7 Two GWASs for systolic (SBP) and diastolic blood pressures (DBP) in ~30 000 individuals of European ancestry identified 13 hypertension-associated loci.2, 3 Subsequently, a large-scale GWAS using a multistage design to examine DBP and SBP in 200 000 individuals of European ancestry detected an additional 16 novel single-nucleotide variants (SNVs).4 However, the latter GWAS detected fewer than 10 SNVs associated with DBP or SBP in non-European populations. This discrepancy could reflect the small sample size or differences in genetic backgrounds among populations, or this finding could be a true and valid result.
For the most part, conventional GWASs have been conducted in a cross-sectional manner that commonly measures traits at a single point in time. SBP, DBP and the prevalence of hypertension are significantly correlated with age. Therefore, control individuals who have normal blood pressure at a certain point will potentially display hypertension after a few years. Conventional cross-sectional GWASs do not consider this possibility. Given that longitudinal GWASs evaluate temporal changes in blood pressure and the prevalence of hypertension related to age, this analysis increases the statistical power to detect these associations. Therefore, we traced disease progression and physiological changes in 6026 Japanese individuals who underwent annual health check-ups for several years. We performed a longitudinal exome-wide association study (EWAS), which is suited to studying the effects of low frequency or rare variants within a gene of interest, to explore novel genetic variants that confer susceptibility to hypertension, focusing on rare coding variants, such as East Asian-specific alleles. The results of this longitudinal EWAS enhance the current understanding of the relationship between ethnic-specific genetic factors and diseases.
Materials and methods
Ethics statement
The study protocol complied with the Declaration of Helsinki and was approved through the Committees on the Ethics of Human Research of Mie University Graduate School of Medicine and Inabe General Hospital. Written informed consent was obtained from all subjects before enrolment in the present study.
Study population
The 6026 community-dwelling individuals in Inabe City, Mie Prefecture, Japan, were recruited from individuals who visited the health-care center of Inabe General Hospital for an annual health check-up and were followed-up yearly (Inabe cohort). We refer to this cohort as the ‘Inabe cohort’. Individuals in the Inabe cohort were registered between March 2010 and September 2012, and genomic DNA was extracted from venous blood cells using a DNA extraction kit (SMITEST EX-R&D; Medical and Biological Laboratories, Nagoya, Japan) and stored in the genomic DNA bank of the Research Center for Genomic Medicine at Mie University. For all participants, medical examination data obtained from April 2003 to March 2014 (11 years) were deposited into a database. Each subject had one set of health data for each year of attendance at the clinic. Therefore, all participants had undergone 1–11 medical examinations (a total of 28 529 examinations), and the average follow-up period was 5 years. The detailed characteristics of the study subjects in a 5-year follow-up are described elsewhere.8
Among the 6026 subjects in the Inabe cohort, 2249 patients were affected by essential hypertension, and the remaining 3777 patients were treated as controls. Hypertension was defined as either DBP of ⩾90 mm Hg or SBP of ⩾140 mm Hg (or both), by virtue of the subject having taken antihypertensive medication. Blood pressure was measured at least twice with the subjects having rested in the sitting position for >5 min. A skilled physician or nurse obtained the measurements according to the guidelines of the American Heart Association.9 The mean DBP of all subjects was 74.7 mm Hg, whereas the mean SBP was 120.7 mm Hg. The mean age of subjects was 52.5 years of age (18–91 years of age) and the mean body mass index was 23.0 kg m−2. The percentages of missing data were 2.2% for DBP, SBP and body mass index. There were no missing data for age, gender or smoking status.
Longitudinal EWAS
A longitudinal EWAS for hypertension was performed on the Inabe cohort, and Infinium HumanExome-12 ver. 1.2 BeadChip or Infinium Exome-24 ver. 1.0 BeadChip (Illumina, San Diego, CA, USA) were used to genotype ~244 000 SNVs. The exome array includes putative functional exonic variants selected from >12 000 ethnically diverse individuals.10 Previously available marker sets of GWASs were designed to identify common alleles (minor allele frequency (MAF) of ⩾5%) and were not well suited to studying the effects of low frequency or rare variants within a gene of interest. The exome arrays used in the present study contained ~244 000 SNVs, including common, low-frequency (0.5%⩽MAF<5%) and rare (MAF<0.5%) variants, although EWAS is a focused genotyping method that differs from GWAS, which includes up to 4.5 million markers for SNVs and copy number variations. Using JMP Genomics version 6.0 (SAS Institute, Cary, NC, USA), the genotyping data of 6026 individuals were converted into binary data with the dominant and recessive models, after removing monomorphic sites among subjects, resulting in 58 563 SNVs for the dominant model and 38 725 SNVs for the recessive model. In a similar manner, the genotyping data were also converted into numerical data with the additive model, generating 58 563 SNVs. The dominant and recessive models were defined as ‘AA (0) vs AB+BB (1)’ and ‘AA+AB (0) vs BB (1)’ (A, major allele; B, minor allele), respectively, whereas the additive model was defined as ‘AA (0)P<0.001) in controls were also discarded.
Sitlani et al.11 reported that a small effective sample size can increase the chances of generating false positives (type I errors). In the preliminary analysis, a MAF of <0.01 appeared to increase type I errors in the recessive model because extraordinarily large numbers of SNVs were identified as hypertension-associated SNVs. Therefore, we discarded SNVs with a MAF of <0.05. Quantile–quantile plots for P-values of allele frequencies in the EWASs for DBP, SBP, or the prevalence of hypertension in all genetic models are shown in Supplementary Figure S1. The genomic inflation factor (λ) of P-values was 1.10 for the prevalence of hypertension and 1.09 for DBP and SBP in the dominant model (Supplementary Figure S1a). In the additive model, the λ was 1.12 for the prevalence of hypertension and 1.09 for DBP and SBP (Supplementary Figure S1b). In the recessive model, the λ was 1.07 for the prevalence of hypertension, 1.09 for DBP and 1.14 for SBP (Supplementary Figure S1c).
The genotype data of participants in the EWAS were examined for population stratification using principal component analysis according to the EIGENSTRAT method12 with the JMP Genomics program (Supplementary Figure S2), and we excluded four outliers identified in the analysis. The rearrangement of Inabe longitudinal data was conducted using R software version 3.3213 via RStudio version 1.0.13614 and Perl script.
Statistical analyses
The association of longitudinal changes in the prevalence of hypertension, DBP and SBP with genetic variants was examined using the generalized estimating equation (GEE) model15, 16 with adjustments for age, gender, body mass index and smoking status, using the R package ‘geepack’.17 The waves argument was used to specify the ordering of repeated measurements within individuals. The statistical significance of the association was P<8.54 × 10−7 (0.05/58 563 SNVs) for dominant and additive models and P<1.29 × 10−6 (0.05/38 725 SNVs) for the recessive model after applying Bonferroni’s correction to compensate for the multiple comparison of genotypes with the clinical parameters. Sitlani and colleagues proposed that compared with the use of normal distribution, the use of a t reference distribution with estimated degrees of freedom for the distributional assumption of the ratio of coefficient estimate to its standard error can improve the accuracy of the significance detected with the GEE. In addition, these authors recommended the use of approxdf, a scale of small effective sample size:11 approxdf=2 × MAF × Nindep, where Nindep is the sum of the estimated number of independent observations per person. We also computed the P-value via GEE using a t reference distribution with Satterthwaite estimates of degrees of freedom and approxdf using the R package ‘bosswithdf’,11, 18 and discarded SNVs with approxdf <10, as approxdf ⩾10 decreases the chances of generating type I errors.11 The GEE with t reference distribution was implemented using the clinical parameters of subjects in the last 5 years for which data were available.
The significance of difference of DBP or SBP between groups of subjects with different genotypes was assessed using Welch’s t-test (two-sided P-value <0.05). Haplotype phase and linkage disequilibrium (LD) among SNVs were estimated using IMPUTE2 version 2.3.219 and Haploview version 4.220 programs, respectively. Perl and R scripts were written to convert the SNV data used in the present study into suitable formats for each program.
Survey of allele frequencies in human populations and variants in nonhuman primates
Information on allele frequencies of target SNVs within human populations was obtained from the 1000 Genomes Project21 (http://www.internationalgenome.org/) and the Integrative Japanese Genome Variation Database (iJGVD) from the Tohoku Medical Megabank Organization of Tohoku University22 (https://ijgvd.megabank.tohoku.ac.jp/author/tommo/). In addition, a genetic variant at the homologous SNVs within vertebrates was investigated in the UCSC Genome Browser (for archaic humans) or Multiz Alignments of 100 Vertebrates in the UCSC database (http://genome.ucsc.edu), and information on allele frequencies in great apes was obtained from the Great Ape Genome Project database23 (http://biologiaevolutiva.org/greatape/).
Results
Association of SNVs with hypertension in the Inabe cohort
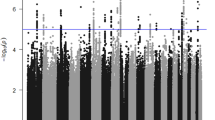
In the present study, the GEE model with adjustments for age, gender, body mass index and smoking status was used to examine the association between 58 563 SNVs for the dominant and additive models, and 38 725 SNVs for the recessive model and clinical parameters related to hypertension in the Inabe cohort (after removing four outliers identified using the principal component analysis; Supplementary Figure S2). In the dominant and additive models, six SNVs located at 12q24.11–12q24.13 were significantly (P<8.54 × 10−7) associated with all three clinical parameters: the prevalence of hypertension; SBP; and DBP (Figure 1; Table 1). In addition, three SNVs located on chromosome 10 were significantly associated with SBP in the dominant and additive models, and one additional SNV located on the same chromosomal region showed a significant association with SBP in the additive model (Figure 1; Table 1). In the recessive model, four SNVs were significantly (P<1.29 × 10−6) associated with the prevalence of hypertension, and one and five SNVs were significantly associated with SBP and DBP, respectively (Figure 1; Table 1).

Single-nucleotide variants (SNVs) showing significant associations with hypertension. The associations were examined using the generalized estimating equation (GEE) model in the dominant (a), additive (b) and recessive (c) genetic models. The seven candidates of hypertension-associated SNVs shown in the figure were determined when the association was supported by the GEE model with normal and t reference distributions, and the approxdf value of these SNVs was higher than 30 (see text). The three GEE analyses independently tested the association between SNVs and three clinical parameters: prevalence of hypertension (HT); systolic blood pressure (SBP); and diastolic blood pressure (DBP). On the basis of Bonferroni’s correction, P-values of <8.54 × 10−7 for the dominant and additive models and P<1.29 × 10−6 for the recessive model were considered statistically significant. aAmino-acid change reflecting a point mutation at each SNV (nonsynonymous substitution).
To confirm the associations between candidate SNVs detected in the longitudinal EWAS and clinical parameters related to hypertension, we applied the GEE model with t reference distribution and threshold of approxdf (⩾10). Additional analysis showed that all six SNVs at 12q24.11–12q24.13 (rs12229654 at 12q24.11, rs3782886 of BRAP, rs11066015 of ACAD10, rs671 of ALDH2, and rs2074356 and rs11066280 of HECTD4) detected in the dominant and additive models and one SNV (rs11917356 of COL6A5) in the recessive model showed significant associations (P<0.05/the number of candidate SNVs in each genetic model) and values of approxdf were >30 (Table 1). Consequently, we identified the seven SNVs as genetic variants that confer susceptibility to hypertension (Table 2).
Identification of East Asian-specific haplotype on chromosome 12
Our longitudinal EWAS for hypertension in the dominant and additive models revealed six significant SNVs. On the basis of the information obtained from the 1000 Genomes Project21 and iJGVD databases, we examined allele frequencies among human populations (Table 3). Interestingly, minor (derived) alleles of all six SNVs were specifically detected in East Asia. This result suggests that the derived alleles were recently expanded throughout East Asia. On the basis of clinical parameters of subjects in the latest year for which data were available, the prevalence of hypertension in subjects with the East Asian-specific allele was lower than that of those with common (ancestral) alleles at each SNV site (mean odds ratio=0.78, P<1.0 × 10−8 using Fisher’s exact test). Among local Japanese populations, the frequencies of the East Asian alleles were highest in the Inabe cohort (Table 3).
The six hypertension-associated SNVs were located at 12q24.1. Therefore, using the SNV data obtained in the present study, we estimated the LD among these SNVs based on 232 biallelic sites spanning an ~2.0 Mb genomic region on chromosome 12 (Supplementary Figure S3). The LD among the six SNVs was relatively strong for the physical distance. Particularly, five SNVs at 12q24.12–12q24.13 (excluding rs12229654 at 12q24.11) showed strong LDs (r2>0.8). Therefore, we used these five SNVs for a haplotype estimate.
Using the IMPUTE2 program, we generated phased haplotypes comprising 232 biallelic sites, and examined the genotype containing the five hypertension-related SNV motifs (from left, rs3782886 of BRAP, rs11066015 of ACAD10, rs671 of ALDH2, and rs2074356 and rs11066280 of HECTD4) for all subjects. In the Inabe cohort, the proportion of diplotype ‘TGGGT/TGGGT’ was the highest (2274 subjects, 46.1%), followed by ‘TGGGT/CAAAA’ (2109 subjects, 35.0%) and ‘TGGGT/CAAAA’ (460 subjects, 7.6%; Supplementary Table S1). The ‘TGGGT’ haplotype comprises all ancestral alleles at each SNV, while the ‘CAAAA’ haplotype comprises all derived alleles.
The haplotype ‘CAAAA’ is East Asian-specific. However, whether this haplotype is specific to the modern human remains unknown. To address this question, we examined putative haplotypes in other vertebrates based on information in the UCSC Genome Browser or Multiz Alignments of 100 Vertebrates in the UCSC database (Figure 2; Supplementary Table S2). No other species with the East Asian-specific haplotype of modern humans were observed and the common haplotype was predominantly observed in Haplorhini, and even archaic humans, including Neanderthal and Denisovan. Notably, a guanine (ancestral allele) at rs671 in ALDH2 appeared to be consistently conserved in all vertebrates, including a jawless vertebrate, sea lamprey (Petromyzon marinus). Therefore, the nonsynonymous substitution at this site might be disadvantageous for survival in vertebrates.

Comparison of five hypertension-associated single-nucleotide variants (SNVs) among primates. The primate allele identical to the human ancestral allele is shown in bold. aThe putative allele was estimated based on the UCSC Genome Browser or Multiz Alignments of 100 Vertebrates in the UCSC database. bThe putative allele was estimated based on allele frequencies deposited in the Great Ape Genome Project database.
To examine genetic polymorphisms within nonhuman primates, variant call format data sets of 38 chimpanzees (Pan troglodytes and Pan paniscus), 31 gorillas (Gorilla gorilla and Gorilla beringei) and 10 orangutans (Pongo pygmaeus and Pongo abelii) were retrieved from the Great Ape Genome Project database.23 We subsequently searched allele frequencies at the counterparts to five human hypertension-related SNVs densely located at 12q24.12–12q24.13. However, the variant call format data sets did not contain any information for the allele frequencies, suggesting that the three primates examined do not have genetic polymorphisms at these sites. Therefore, it is likely that the East Asian-specific haplotype identified in the present study is human-specific.
Comparison of clinical parameters between East Asian-specific and common haplotypes
In the Inabe cohort, the prevalence of hypertension in subjects with the East Asian haplotype (case=1052 and control=2111) was significantly lower than in subjects with the common haplotype (case=4875 and control=3141; odds ratio=0.32, P<2.2 × 10−16 by Fisher’s exact test). The odds ratio was lower than that of each SNV (the mean odds ratio was 0.78, see above), suggesting that the effect of haplotype on hypertension is greater than that of individual SNVs. In the combined group of homozygotes with the East Asian haplotype (CAAAA/CAAAA) and heterozygotes with East Asian and common haplotypes (CAAAA/TGGGT), the mean SBP and DBP values were 119.6±0.32 and 73.6±0.24, respectively. These values were significantly lower than the mean SBP (121.5±0.31) and DBP (75.5±0.24) values of homozygotes with the common haplotype (P<1.8 × 10−5 and <1.6 × 10−8 for SBP and DBP, respectively, using Welch’s t-test), according to cross-sectional analysis using clinical parameters of subjects in the latest year for which data were available. These results suggest that the East Asian haplotype is protective against hypertension.
To survey the effect of different lifestyle variables (drinking, smoking and exercise status) on hypertension among diplotypes, we compared the SBP and DBP values of each diplotype using cross-sectional analysis data in a pairwise fashion (for example, smokers and non-smokers). The mean SBP and DBP values of smokers and drinkers were significantly higher than those of non-smokers and non-drinkers among subjects homozygous for the common diplotypes (Supplementary Figure S4a–d). Moreover, the comparison of DBP values in heterozygotes with the common and East Asian haplotypes showed significant differences between groups based on smoking or drinking status. The differences in the DBP values of smokers and non-smokers with East Asian haplotypes were smaller than those in subjects with a common haplotype. Stratification based on smoking status revealed the degree of difference between the two groups with the ‘CAAAA/CAAAA’ diplotype was 0.35 mm Hg, and the degree of difference between the two groups with ‘TGGGT/CAAAA’ and ‘TGGGT/TGGGT’ diplotypes was 3.01 and 4.33 mm Hg, respectively (Supplementary Figure S4b). This finding suggests that compared with the common haplotype, the East Asian-specific haplotype is less susceptible to the adverse effects of smoking. Using all six detected SNVs in the dominant model, we also estimated phased haplotypes and repeated the stratification analysis. Similar results were observed in analyses based on six SNVs (including rs12229654 at 12q24.11; Supplementary Figure S5a–d).
Identification of a novel genetic variant in the COL6A5 gene
For the recessive model, one SNV (rs11917356) in COL6A5 showed significant association with SBP in the GEE model with normal and t reference distributions, and the value of approxdf was 60.3. We compared the mean SBP and DBP values between genotypes for the candidate SNV. SBP and DBP values in subjects with the GG or GA genotype (mean SBP=120.9±0.22 and mean DBP=74.8±0.17) were significantly higher than those in subjects with the AA genotype (mean SBP=117.7±0.64 and mean DBP=72.7±0.48; P=2.4 × 10−6 and 5.1 × 10−5 for mean SBP and mean DBP, respectively, by Welch’s t-test). This finding suggests that the ‘A’ allele is protective against hypertension.
On the basis of the information obtained from the 1000 Genomes Project21 and iJGVD22 databases, we examined allele frequencies of focal SNVs within human populations (Table 3). Remarkably, the frequency of the derived allele ‘G’ at rs11917356 was highest in Japan (~70%), and the ‘G’ allele was major in East Asian populations and minor in non-East Asian populations, suggesting that the frequencies of the derived alleles have rapidly increased since the split of East Asian and South Asian lineages.
Discussion
In the present longitudinal EWAS, the GEE model with normal and t reference distributions demonstrated that six SNVs located on chromosome 12 were significantly associated with the clinical parameters of hypertension, while one SNV of COL6A5 on chromosome 3 was significantly related to SBP. The effective sample size based on approxdf of these SNVs was >30. In a CHAGE multistudy meta-analysis, the GEE model using t reference distribution with approxdf ⩾10 provided more accurate P-values than standard GEE models.11 Therefore, the reliability of associations between the clinical parameters of hypertension and the candidate SNVs detected in the present study was high.
On the basis of the data for the six candidate SNVs on chromosome 12, we showed that East Asian-specific alleles (or haplotype) could be protective against hypertension. Among the six SNVs, a nonsynonymous nucleotide substitution at rs671 in ALDH2 altered an amino-acid residue (Table 2). However, all nucleotide substitutions in the other SNVs were silent. Therefore, the SNV in ALDH2 may be an important factor in the onset of hypertension. ALDH2 is a mitochondrial acetaldehyde dehydrogenase involved in the major pathway of alcohol metabolism. The rs671 G to A transition in ALDH2 exon 12 changes the amino-acid at position 504 (E504K), resulting in defective enzyme activity.24, 25 Previous studies reported that the derived ALDH2 allele (A) was predominantly observed in East Asians, and this allele has potentially expanded in this area through natural selection in the recent past.26, 27 A meta-analysis of GWASs for blood pressure variation in East Asian populations detected significant associations between rs11066280 of HECTD4 and SBP or DBP values, and an estimated haplotype comprising eight SNVs showed relatively high LD (r2>0.7). Furthermore, haplotypes comprising five of the eight SNVs were clearly differentiated into high- and low-blood-pressure groups.5 In the present longitudinal EWAS, four of the five SNVs detected in the previous study (rs3782886 of BRAP, rs671 of ALDH2, and rs2074356 and rs11066280 of HECTD4) were significantly associated with hypertension. There was high LD among SNVs densely located at 12q24.1, suggesting that positive selection, acting on the site of rs671 in ALDH2, alters the allelic frequency of the other five SNVs through genetic hitchhiking. Indeed, a signal indicative of a selective sweep (recent positive selection) was identified at 12q24.13 near the ALDH2 in two haplotype-based tests.5 Nevertheless, in the present study, the ancestral alleles at the six detected SNVs appeared to be conserved during the course of primate evolution. In addition, the protective effect of the East Asian haplotype on hypertension (the odds ratio=0.3) was greater than that of each East Asian allele (the mean odds ratio=0.8). Therefore, the effect of genetic factors other than rs671 remains controversial. It is intriguing to determine how and why the haplotype generated by unusual alleles at each SNV has spread in East Asians.
As rs671 of ALDH2 influences alcohol intake or consumption, the drinking status of subjects is potentially associated with hypertension. Cross-sectional analysis using the clinical parameters for subjects in the latest year for which data were available revealed that the prevalence of hypertension in non-drinkers with the East Asian allele at rs671 was significantly lower than that in non-drinkers with the common allele (P=0.009 by Fisher’s exact test, odds ratio=0.87). To further consider the effect of alcohol intake and consumption, we re-analyzed the relationship between longitudinal changes in the prevalence of hypertension, SBP, or DBP values and six detected SNVs in the dominant model using the GEE method after adjustment for alcohol intake or alcohol consumption (also including age, gender, body mass index and smoking status; Supplementary Table S3). The associations of all six SNVs with clinical parameters of hypertension were supported by the results of the GEE test, suggesting that the identified genetic variants at 12q24.1 could influence susceptibility to hypertension.
Given that their genetic variations influence the incidence of hypertension, it is unclear how these variants are involved in the pathogenesis of hypertension. While the immediate cause is unknown, differences in sodium retention might be associated with hypertension. Hence, we preliminarily analyzed the interaction between molecules encoded by genes with the five hypertension-associated SNVs and molecules directly or indirectly related to the regulation of sodium retention (such as RAC1, ADRB2, WNK4, NR3C2, SLC12A3, SCNN1A and SCNN1B). To perform this analysis, we used Cytoscape version 3.4.028 software, which automatically integrates human molecular interaction data from different public databases (Supplementary Figure S6). The integrated network did not show any direct interaction among the analyzed molecules. However, the network displayed some potential indirect interactions. For example, HECTD4 was associated with YWHAZ, which is involved in the regulation of signaling pathways. Meek et al.29 previously showed the physical association of these molecules. According to the molecular INTeraction database (http://mint.bio.uniroma2.it), Pozuelo-Rubio et al.30 reported the association of YWHAZ and WNK4, a regulator of renal electrolyte transport, using tandem affinity purification. WNK4 has an important role in blood pressure regulation.31 In addition, in the generated network, ALDH2 was indirectly associated with the WNK4 via CUL3, a component of ubiquitin E3 ligase, according to the Biological General Repository for Interaction Data sets (https://thebiogrid.org/). These results are not direct evidence that the four hypertension susceptibility genes identified at 12q24.1 in the present study influence the sodium retention ability, and further analyses are required to reveal the pathogenesis of hypertension.
Comparison of the DBP values between smokers and non-smokers in each diplotype of phased haplotypes (East Asian and common haplotypes) showed a clear pattern. The degree of differences of DBP values between the two groups defined according to smoking status decreased with the number of East Asian haplotypes in the diplotype. These data were derived from the cross-sectional analysis using clinical parameters of subjects in the latest year for which data were available. Cigarette smoking potentially increases the risk of cardiovascular diseases.32, 33 Nevertheless, in the present study, subjects with the East Asian haplotype were less susceptible to the adverse effects of smoking on hypertension compared with those with the common haplotype. Thus, the interaction between the genetic factor of common haplotype and the environmental factor of smoking may intensify the adverse effects on hypertension.
The longitudinal EWAS for the recessive model showed one candidate SNV in COL6A5. According to information on GWAS central (http://www.gwascentral.org/) and GWAS catalog (https://www.ebi.ac.uk/gwas/) databases, rs11917356 is a novel genetic variant associated with hypertension. In addition, the frequency of derived allele ‘G’ was remarkably higher in East Asia (particularly in Japan) than in the other ethnic populations. This finding suggests that the derived allele rapidly spread throughout East Asia. However, the mean SBP and DBP values in subjects with the derived allele were significantly higher than those in subjects with the ancestral allele ‘A’. Given that the ‘G’ allele in Japanese individuals could be susceptible to hypertension, it remains unclear why the frequency of the allele has increased in East Asia. It is possible that the derived allele confers adaptability to environments in East Asia in return for the risk of hypertension incidence.
COL6A5 (collagen type VI alpha 5 chain) is a member of the collagen superfamily. A previous study reported that COL6A5 is a susceptibility gene for the chronic inflammatory skin disorder atopic dermatitis.34 The rs11917356 represents a G>A transition that alters the amino-acid residue at position 982 (D982G), but this substitution is unlikely to have a strong impact on protein function, according to SIFT (http://sift.jcvi.org/, score=0.73) and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/index.shtml, score=0.093). The COL6A5 gene is primarily expressed in the skin but is also expressed in the lungs, small intestine, colon and testes.34 In addition, COL6A5 protein is strongly expressed around blood vessels at the interface between the papillary and reticular dermis.35 Thus, the amino-acid change in COL6A5 may be associated with hypertension through changes in blood vessel elasticity (or stiffness). Although the pathogenesis of hypertension resulting from genetic variation at rs11917356 remains unclear, the results of the present study suggest that the genetic variant of COL6A5 confers susceptibility to hypertension.
There were certain limitations in the present study. First, the longitudinal EWAS was conducted only in a local Japanese population. Given that the subjects were community-dwelling individuals who visited the health-care center for an annual health check-up, the selection bias was small in the present study. Although multicenter longitudinal EWASs improved the accuracy and reliability of these results, those data are not currently available. Therefore, the replication of longitudinal EWASs in other Japanese populations or other ethnic groups is required to clarify the associations of the identified SNVs with hypertension. Second, the functional relevance of candidate SNVs identified in the longitudinal EWAS to the pathogenesis of hypertension remains unclear. Third, the follow-up period of annual health check-ups varied from 1 to 11 years among individuals.
In conclusion, the results of the present study suggest that the East Asian-specific haplotype, comprising SNVs genotyped on 12q24.1 and a COL6A5 ancestral allele of which the frequency may have recently decreased in East Asians, may represent protective genes against essential hypertension. In addition, the East Asian-specific haplotype may be less susceptible to the adverse effects of smoking on hypertension. Clinical genotyping for the SNVs detected in this longitudinal EWAS may be useful for precise and/or personalized medicine.
References
Ministry of Health, Labour and Welfare Patient Survey 2016, Available at http://www.mhlw.go.jp/english/database/db-hss/ accessed 4 January 2017.
Newton-Cheh C, Johnson T, Gateva V, Tobin MD, Bochud M, Coin L et al. Genome-wide association study identifies eight loci associated with blood pressure. Nat Genet 2009; 41: 666–676.
Levy D, Ehret GB, Rice K, Verwoert GC, Launer LJ, Dehghan A et al. Genome-wide association study of blood pressure and hypertension. Nat Genet 2009; 41: 677–687.
International Consortium for Blood Pressure Genome-Wide Association Studies. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature 2011; 478: 103–109.
Kato N, Takeuchi F, Tabara Y, Kelly TN, Go MJ, Sim X et al. Meta-analysis of genome-wide association studies identifies common variants associated with blood pressure variation in east Asians. Nat Genet 2011; 43: 531–538.
Cho YS, Go MJ, Kim YJ, Heo JY, Oh JH, Ban H-J et al. A large-scale genome-wide association study of Asian populations uncovers genetic factors influencing eight quantitative traits. Nat Genet 2009; 41: 527–534.
Simino J, Shi G, Bis JC, Chasman DI, Ehret GB, Gu X et al. Gene-age interactions in blood pressure regulation: a large-scale investigation with the CHARGE, global BPgen, and ICBP consortia. Am J Hum Genet 2014; 95: 24–38.
Yamada Y, Matsui K, Takeuchi I, Oguri M, Fujimaki T . Association of genetic variants with hypertension in a longitudinal population-based genetic epidemiological study. Int J Mol Med 2015; 35: 1189–1198.
Perloff D, Grim C, Flack J, Frohlich ED, Hill M, McDonald M et al. Human blood pressure determination by sphygmomanometry. Circulation 1993; 88: 2460–2470.
Grove ML, Yu B, Cochran BJ, Haritunians T, Bis JC, Taylor KD et al. Best Practices and Joint Calling of the HumanExome BeadChip: The CHARGE Consortium. PLoS ONE 2013; 8: e68095.
Sitlani CM, Rice KM, Lumley T, McKnight B, Cupples LA, Avery CL et al. Generalized estimating equations for genome-wide association studies using longitudinal phenotype data. Stat Med 2015; 34: 118–130.
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D . Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006; 38: 904–909.
R Core Team R: A language and environment for statistical computing 2016 Available at http://www.r-project.org/ [21 Dec. 2016].
RStudio Team RStudio: Integrated Development Environment for R. RStudio, Inc. 2015, Available at http://www.rstudio.com/ [8 Jan. 2017].
Liang KY, Zeger SL . Longitudinal data analysis using generalized linear models. Biometrika 1986; 73: 13–22.
Hanley JA, Negassa A, Edwardes MD, Forrester JE . Statistical analysis of correlated data using generalized estimating equations: an orientation. Am J Epidemiol 2003; 157: 364–375.
Halekoh U, Højsgaard S, Yan J . The R package geepack for generalized estimating equations. J Stat Softw 2006; 15: 1–11.
Voorman A, Rice K, Lumley T . Fast computation for genome-wide association studies using boosted one-step statistics. Bioinformatics 2012; 28: 1818–1822.
Howie BN, Donnelly P, Marchini J, Rioux J, Xavier R, Taylor K et al. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 2009; 5: e1000529.
Barrett JC, Fry B, Maller J, Daly MJ . Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005; 21: 263–265.
The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature 2010; 467: 1061–1073.
Nagasaki M, Yasuda J, Katsuoka F, Nariai N, Kojima K, Kawai Y et al. Rare variant discovery by deep whole-genome sequencing of 1070 Japanese individuals. Nat Commun 2015; 6: 8018.
Prado-Martinez J, Sudmant PH, Kidd JM, Li H, Kelley JL, Lorente-Galdos B et al. Great ape genetic diversity and population history. Nature 2013; 499: 471–475.
Hsu LC, Bendel RE, Yoshida A . Direct detection of usual and atypical alleles on the human aldehyde dehydrogenase-2 (ALDH2 locus. Am J Hum Genet 1987; 41: 996–1001.
Crabb DW, Edenberg HJ, Bosron WF, Li TK . Genotypes for aldehyde dehydrogenase deficiency and alcohol sensitivity. The inactive ALDH22 allele is dominant. J Clin Invest 1989; 83: 314–316.
Oota H, Pakstis AJ, Bonne-Tamir B, Goldman D, Grigorenko E, Kajuna SLB et al. The evolution and population genetics of the ALDH2 locus: random genetic drift, selection, and low levels of recombination. Ann Hum Genet 2004; 68: 93–109.
Luo HR, Wu GS, Pakstis AJ, Tong L, Oota H, Kidd KK et al. Origin and dispersal of atypical aldehyde dehydrogenase ALDH2*487Lys. Gene 2009; 435: 96–103.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003; 13: 2498–2504.
Meek SEM, Lane WS, Piwnica-Worms H . Comprehensive proteomic analysis of interphase and mitotic 14-3-3-binding proteins. J Biol Chem 2004; 279: 32046–32054.
Pozuelo-Rubio M . Proteomic and biochemical analysis of 14-3-3-binding proteins during C2-ceramide-induced apoptosis. FEBS J 2010; 277: 3321–3342.
Wu G, Peng JB . Disease-causing mutations in KLHL3 impair its effect on WNK4 degradation. FEBS Lett 2013; 587: 1717–1722.
Najem B, Houssière A, Pathak A, Janssen C, Lemogoum D, Xhaët O et al. Acute cardiovascular and sympathetic effects of nicotine replacement therapy. Hypertension 2006; 47: 1162.
Bowman TS, Gaziano JM, Buring JE, Sesso HD . A prospective study of cigarette smoking and risk of incident hypertension in women. J Am Coll Cardiol 2007; 50: 2085–2092.
Söderhäll C, Marenholz I, Kerscher T, Rüschendorf F, Esparza-Gordillo J, Worm M et al. Variants in a novel epidermal collagen gene (COL29A1 are associated with atopic dermatitis. PLoS Biol 2007; 5: 1952–1961.
Sabatelli P, Gara SK, Grumati P, Urciuolo A, Gualandi F, Curci R et al. Expression of the collagen VI α5 and α6 chains in normal human skin and in skin of patients with collagen VI-related myopathies. J Invest Dermatol 2011; 131: 99–107.
Acknowledgements
This work was supported by the Kurata Grant awarded by the Hitachi Global Foundation (to Y Yasukochi and Y Yamada); CREST (H25-H30) of the Japan Science and Technology Agency (to Y Yamada, JS and IT); and the Japan Society for the Promotion of Science KAKENHI grants (JP15H04772 to Y Yasukochi and JP15H04772 to Y Yamada).
Publisher’s note:
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Experimental & Molecular Medicine website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Yasukochi, Y., Sakuma, J., Takeuchi, I. et al. Longitudinal exome-wide association study to identify genetic susceptibility loci for hypertension in a Japanese population. Exp Mol Med 49, e409 (2017). https://doi.org/10.1038/emm.2017.209
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2017.209
This article is cited by
-
Cold-induced vasodilation response in a Japanese cohort: insights from cold-water immersion and genome-wide association studies
Journal of Physiological Anthropology (2023)
-
Positive selection acts on regulatory genetic variants in populations of European ancestry that affect ALDH2 gene expression
Scientific Reports (2022)
-
Collagen type VI α5 gene variations may predict the risk of lung cancer development in Chinese Han population
Scientific Reports (2020)
-
Genomics of disease risk in globally diverse populations
Nature Reviews Genetics (2019)
