
Abstract
Diabetic retinopathy (DR), one of the most serious complications of diabetes, has been associated with inflammatory processes. We have recently reported that interleukin (IL)-17A, a proinflammatory cytokine, is increased in the plasma of diabetic patients. Further investigation is required to clarify the role of IL-17A in DR. Ins2Akita (Akita) diabetic mice and high-glucose (HG)-treated primary Müller cells were used to mimic DR-like pathology. Diabetes induced retinal expression of IL-17A and IL-17 receptor A (IL-17RA) in Müller cells in contrast to ganglion cells. Further evidence demonstrated that retinal Müller cells cultured in vitro increased IL-17A and IL-17RA expression as well as IL-17A secretion in the HG condition. In both the HG-treated Müller cells and Akita mouse retina, the Act1/TRAF6/IKK/NF-κB signaling pathway was activated. IL-17A further enhanced inflammatory signaling activation, whereas Act1 knockdown or IKK inhibition blocked the downstream signaling activation by IL-17A. HG- and diabetes-induced Müller cell activation and dysfunction, as determined by increased glial fibrillary acidic protein, vascular endothelial growth factor and glutamate levels and decreased glutamine synthetase and excitatory amino acid transporter-1 expression, were exacerbated by IL-17A; however, they were alleviated by Act1 knockdown or IKK inhibition. In addition, IL-17A intravitreal injection aggravated diabetes-induced retinal vascular leukostasis, vascular leakage and ganglion cell apoptosis, whereas Act1 silencing or anti-IL-17A monoclonal antibody ameliorated the retinal vascular damage and neuronal cell apoptosis. These findings establish that IL-17A exacerbates DR-like pathology by the promotion of Müller cell functional impairment via Act1 signaling.
Similar content being viewed by others

Introduction
Diabetic retinopathy (DR), which represents one of the most serious complications of diabetes, is a leading cause of blindness in working-age individuals worldwide.1 The pathogenesis of DR is complex, and several vascular, inflammatory and neuronal mechanisms are involved.2 The involvement of inflammatory processes in the induction of structural and functional alterations associated with DR is gaining increasing attention, and it has been particularly associated with the early stages of DR.3, 4 Recent studies have demonstrated that levels of proinflammatory cytokines, including tumor necrosis factor-α, interleukin (IL)-1β, IL-6 and IL-8, are substantially increased in the ocular fluid of diabetic patients.5, 6, 7 The inflammatory components contribute to the pathological changes in DR, including blood–retinal barrier (BRB) breakdown, retinal neovascularization, retinal glutamate metabolic dysfunction and retinal neuronal apoptosis.8, 9, 10, 11 However, the present evidence regarding inflammatory events in DR is restricted to the involvement of innate immunity driven by retinal macrophages and microglia. The involvement of adaptive immune cells, particularly T cells, in DR has not been elucidated.
T helper type 17 (Th17) cells, a subset of CD4+ T cells, produce IL-17 that is currently recognized as a family of cytokines that includes IL-17A, IL-17B, IL-17C, IL-17D, IL-17E and IL-17F.12, 13 The most widely investigated cytokine of this family, IL-17A, is a proinflammatory cytokine and has been implicated in the pathogenesis of several autoimmune and inflammatory diseases. However, limited studies indicate a disorder in IL-17A that may have a significant impact on the course of autoimmune diabetes.14 The limited available studies demonstrate increased secretion of IL-17A from activated peripheral blood T cells in children with type 1 diabetes.15 We have also recently reported that the plasma IL-17A level is increased in type 2 diabetic patients.16 Further investigation is required to clarify the role of IL-17A in DR. In addition, the identification of the IL-17A signaling cascade involved in DR is meaningful for the potential application of IL-17A signaling antagonists to remedy DR. IL-17A signals through a heterodimeric receptor complex that consists of IL-17RA and IL-17RC that recruits nuclear factor (NF)-κB activator 1 (Act1) as an adaptor molecule for downstream signaling; in turn, the IL-17RA/C-Act1 complex triggers tumor necrosis factor receptor-associated factor-6 (TRAF6)-dependent inhibitor of NF-κB kinase (IKK) activation that results in NF-κB activation and ultimately target gene transcription.17, 18
Müller cells, the specific glial cells that span approximately the entire thickness of the retina from the outer limiting membrane to the inner limiting membrane, play an essential role in preserving normal retinal function via the maintenance of homeostasis of the retinal extracellular environment.19, 20 Müller cells participate in the formation of BRB, regulate retinal glutamate metabolism and support retinal neuronal survival.21, 22, 23 Müller cell activation, as demonstrated by an upregulation of glial fibrillary acidic protein (GFAP), has been identified in the retinas of both diabetic patients and diabetic rodents in the early stages of DR.24, 25, 26 Müller cell-derived vascular endothelial growth factor (VEGF) accumulation results in diabetic retinal neovascularization and vascular leakage.27 Hyperglycemia-induced decreases in both glutamine synthetase (GS) and excitatory amino acid transporter-1 (EAAT1) in Müller cells may trigger apoptosis of retinal ganglion cells in DR because of excitotoxicity caused by excessive glutamate.28, 29 Thus, we hypothesized that Müller cells represent a pivotal target of IL-17A in the DR process, in which IL-17A exacerbates BRB breakdown and retinal neuronal cell apoptosis, the major characteristics of DR. This hypothesis was investigated in the current study using high-glucose (HG)-treated primary retinal Müller cells and diabetic model mice with DR.
Materials and methods
Animals
Male heterozygous Ins2Akita (Akita) mice (Stock No. 003548 in Jackson Laboratory, Bar Harbor, ME, USA) with a C57BL/6 background and age-matched nondiabetic siblings were purchased from the Model Animal Research Center of Nanjing University (Nanjing, China). As a result of a spontaneous mutation of the insulin 2 gene, Akita mice develop insulin-dependent diabetes (diabetes mellitus type 1), which is characterized by robust hyperglycemia, at the age of 4 weeks.10, 30 The mice were subjected to intravitreal injection or were killed via a lethal dose of intraperitoneal sodium pentobarbitone (150 mg kg−1 body weight) three months after the onset of diabetes; this approach was based on a previous report that at 10–14 weeks after hyperglycemia in Akita mice, the retinas exhibit evident DR pathology, such as vascular leakage, neuronal cell death and leukostasis.31 All procedures were conducted in compliance with the Association for Research in Vision and Ophthalmology (ARVO) Statement for the Use of Animals in Ophthalmic and Vision Research and were approved by the Institutional Animal Care and Use Committees of Nanjing Medical University.
Intravitreal injection
Under a surgical microscope, a 30-gauge puncture was performed 1 mm behind the corneoscleral limbus in mice that had been deeply anesthetized by isoflurane inhalation; a 33-gauge needle mounted on a 10-μl microsyringe (Hamilton, Reno, NE, USA) was subsequently inserted through the puncture with a 45° angle into the vitreous cavity.32, 33 Mouse recombinant IL-17A (8 or 40 ng per 2 μl in phosphate-buffered saline (PBS) per eye; BioVision, Milpitas, CA, USA) or anti-IL-17A-neutralizing monoclonal antibody (mAb; 2 or 10 μg per 2 μl in PBS per eye; R&D Systems, Minneapolis, MN, USA) were injected into the vitreous cavity of the Akita mice. An adenoviral vector that expressed short hairpin RNA targeting Act1 (Ad-Act1-shRNA) was constructed, amplified and purified by Hanbio Biotechnology in Shanghai, China. Ad-Act1-shRNA was delivered into the vitreous cavity of the Akita mice (2 μl per eye that contained 2 × 108 viral particles). An equivalent volume of adenoviral vector that expressed green fluorescent protein (Ad-GFP) served as the control. The mice that received an intravitreal adenoviral injection or that were used for the retinal angiography were killed 2 weeks after the intravitreal injection, whereas the remaining mice were killed 2 days after the intravitreal injection.
Cell culture and treatments
Primary cultures of rat retinal Müller cells were prepared as previously described with modifications.29 The dissociated retinal tissue of rats at postnatal days 5 to 7 was seeded in culture dishes that contained Dulbecco’s modified Eagle’s medium supplemented with 5 mM D-glucose (normal glucose as a control). The cultures were forcibly pipetted every 3 days to purify the Müller cell population until the percentage of cultured Müller cells, which was identified by the GS expression, exceeded 98%.34 The cells were maintained in Dulbecco’s modified Eagle’s medium with 25 mM D-glucose, as a HG treatment, for 48 h to mimic diabetes in vitro. IL-17A (25 ng ml−1) and/or the IKK inhibitor Wedelolactone (Wedel; 10 μM; Sigma-Aldrich, St Louis, MO, USA) were applied to the primary Müller cell cultures for 24 h in the HG condition. Ad-Act1-shRNA at a multiplicity of infection of 20 infected primary Müller cells in the HG medium, and Ad-GFP was used as a control. After infection for 24 h, the adenoviruses were removed, and the cells were cultured for an additional 24 h before harvest.
Western blot analysis
Cells and retinas were isolated and homogenized in radio immunoprecipitation assay buffer. Western blot assays were performed according to previously described methods.35 The following primary antibodies were used: IL-17A (1:200; Santa Cruz Biotechnology, Dallas, TX, USA); Act1 (1:200; OriGene Technologies, Rockville, MD, USA); p-p65 and p65 (both 1:500; Cell Signaling, Danvers, MA, USA); IL-17RA (1:200), GFAP (1:10 000), VEGF (1:1000), GS (1:1000), EAAT1 (1:1000), TRAF6 (1:2000), caspase-3 (1:1000) and β-actin (1:5000; all from Abcam, Cambridge, MA, USA). Following incubation with the corresponding secondary antibodies (1:5000; Rockland, Limerick, PA, USA), the membranes were scanned, and the protein band intensities were quantified by Odyssey (LI-COR, Lincoln, NE, USA).
Enzyme-linked immunosorbent assay (ELISA)
Samples were prepared according to the manufacturer’s instructions and the report of Huang et al.10 The GFAP levels expressed in Müller cells, VEGF secreted by Müller cells and IL-17A in retinal lysates and Müller cell culture supernatants were measured using ELISA kits (Millipore, Temecula, CA, USA or R&D Systems).
High-performance liquid chromatography
Before performing high-performance liquid chromatography as previously described,36 the cells and retinas were homogenized in perchloric acid at 4 °C. The glutamate concentrations were calculated by the peak area ratio, and quantification was performed by running standard amino acid solutions under the same conditions.
Retinal vascular leukostasis
The mice anesthetized by isoflurane inhalation were perfused with 10 ml PBS from the left heart ventricle to remove erythrocytes and nonadherent leukocytes. To label adherent leukocytes, the mice were perfused with fluorescein isothiocyanate (FITC)-conjugated concanavalin A (20 μg ml−1 in PBS; 5 mg kg−1 body weight; Vector Laboratories, Burlingame, CA, USA), followed by 10 ml PBS perfusion for flushing out unbound concanavalin A. After the eyes were fixed in 4% paraformaldehyde for 1 h, the retinas were flat mounted and the total number of adherent leukocytes per retina was counted under a fluorescence microscope.
Retinal angiography
The mice were anesthetized via isoflurane inhalation and perfused from the left heart ventricle with 1 ml of PBS that contained 50 mg ml−1 FITC–dextran (2 × 106 molecular weight; Sigma-Aldrich) as described by Han et al.37 The eyes were enucleated 5 min after the perfusion of FITC–dextran and were fixed in 4% paraformaldehyde for 1 h. The retinas were dissected, flat mounted and viewed via fluorescence microscopy.
Immunohistochemistry and TUNEL staining
To prepare frozen retinal sections, the ocular anterior segments were removed, and the eyecups were fixed with 4% paraformaldehyde, followed by cryoprotection in a series of sucrose solutions (10–30%). The sectioned retinas were independently incubated with rabbit antibodies against IL-17A (1:100; Santa Cruz Biotechnology) and IL-17RA (1:100), VEGF (1:200) and active caspase-3 (1:200; all three from Abcam) overnight at 4 °C. For double labeling, the sections were incubated with mouse antibodies against NeuN or GS (both 1:200; Abcam) at 4 °C overnight. The sections were stained with the appropriate secondary antibodies: anti-rabbit Alexa Fluor 647, anti-mouse FITC or anti-mouse Alexa Fluor 647 (1:200; Jackson ImmunoResearch Laboratories, West Grove, PA, USA) for 2 h at room temperature, followed by 4′,6-diamidino-2-phenylindole (DAPI) staining (1:1000, 5 min; Solarbio Science & Technology, Beijing, China). To identify the apoptosis of retinal ganglion cells, the retinal sections labeled with NeuN were stained via TUNEL (TdT-mediated dUTP nick end labeling) according to the manufacturer’s protocol (Roche Diagnostics, Indianapolis, IN, USA). All sections were visualized by confocal microscopy, and the number of double-labeled apoptotic cells was counted. For each retina, three sections were counted and an average was obtained; in each section, three visual fields near the retinal central area were counted as a sum.
Statistical analysis
All data are expressed as the mean±s.d. Statistical analyses were performed with Statistics Package for Social Science (SPSS, 12.0; SPSS, Chicago, IL, USA). Comparisons between two groups were assessed using Student’s t-tests. Multiple comparisons among the groups were evaluated by one-way analysis of variance followed by post hoc analysis. The results were considered statistically significant at P<0.05.
Results
Expression of IL-17A and IL-17RA is upregulated in retinal Müller cells of Akita diabetic model mice
At 3 months after the onset of diabetes, the Akita mice exhibited upregulated IL-17A and IL-17RA expression (Figure 1a) as well as an increased IL-17A content (Figure 1b) in the retina compared with the wild-type (WT) mice. Furthermore, immunohistochemistry indicated that the expression of GS, a Müller cell marker in the retina,38 was decreased; however, both IL-17A and IL-17RA expression was increased in the retinas of the Akita mice compared with the WT mice (Figure 1c). Importantly, in the retinas of the Akita mice, cells that coexpressed GS and IL-17A or GS and IL-17RA were identified; however, the colocalization of NeuN, a marker of neurons, and IL-17A or NeuN and IL-17RA was not identified (Figure 1c).

IL-17A and IL-17RA expression is upregulated in Akita mouse retina and HG-treated Müller cells. At 3 months after the onset of diabetes, the mice were killed, and the retinas were removed for analysis via western blot (a), ELISA (b) and fluorescent immunohistochemistry (c). Immunofluorescent staining on retinal cross-sections indicates that GS-labeled Müller cells cover approximately the entire retinal thickness, and they express IL-17A and IL-17RA in Akita mice, indicated by the arrows. The ganglion cells labeled by NeuN in the retinal GCL did not express IL-17A or IL-17RA. (d) Primarily cultured rat retinal Müller cells, which were labeled by GS, exceeded 98% in the cell purity. (e) IL-17A expression in primarily cultured Müller cells. (f) IL-17A concentration in Müller cell culture supernatants. **P<0.01 vs wild-type (WT) mice or control. ELISA, enzyme-linked immunosorbent assay; GCL, ganglion cell layer; GS, glutamine synthetase; HG, high glucose; IL, interleukin; INL, inner nuclear layer; ONL, outer nuclear layer.
To demonstrate that retinal Müller cells produce IL-17A, we assessed the expression and secretion of IL-17A in primary retinal Müller cells, the purity of which exceeded 98% (Figure 1d). Retinal Müller cells cultured in vitro expressed and secreted IL-17A, and the expression and secretion of IL-17A by Müller cells were increased in the HG condition (Figures 1e and f).
Act1/TRAF6/IKK/NF-κB signaling is enhanced in both HG-treated retinal Müller cells and Akita mouse retinal tissue and further enhanced by IL-17A treatment
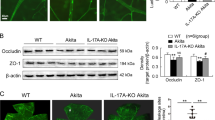
In primary retinal Müller cells, HG induced IL-17RA, Act1 and TRAF6 expression, as well as p65 phosphorylation, and IL-17A further enhanced the HG-induced signaling upregulation (Figure 2a). Ad-Act1-shRNA treatment abolished the IL-17A effects of enhancing Act1 and TRAF6 expression and p65 phosphorylation as well as reduced the HG-induced Act1 and TRAF6 expression and p65 phosphorylation (Figure 2a). Wedel, an inhibitor of IKK, blocked both the IL-17A and HG effects of increased p65 phosphorylation (Figure 2a). As a control for Ad-Act1-shRNA treatment, Ad-GFP treatment did not significantly alter the IL-17A-induced signaling upregulation (Figure 2a).

IL-17A further activates the inflammatory signaling pathway Act1/TRAF6/IKK/NF-κB in both HG-treated retinal Müller cells and Akita mouse retinal tissue. (a) Expression of IL-17A-related signaling molecules in primarily cultured retinal Müller cells. Retinal Müller cells were exposed to HG (25 mM) for 24 h and subsequently treated with IL-17A (25 ng ml−1) and/or the IKK inhibitor Wedel (10 μM) for 24 h. For silencing the Act1 gene, Müller cells were infected with Ad-Act1-shRNA (or Ad-GFP as a control) for 24 h in the HG condition; the adenoviruses were subsequently removed, and the cells were cultured for an additional 24 h. Following the treatments, the proteins of the cells were extracted for western blot analysis. (b) Expression of IL-17RA downstream signaling molecules in the retinas. At 3 months after the onset of diabetes, Akita mice were subjected to intravitreal injection of IL-17A (8 or 40 ng per 2 μl in PBS per eye) or Ad-Act1-shRNA (or Ad-GFP as a control). At 2 days after the IL-17A intravitreal injection or 2 weeks after the Ad-Act1-shRNA intravitreal injection, the proteins of the retinas were extracted for western blot analysis. **P<0.01 vs control or wild type (WT); +P<0.05, ++P<0.01 vs HG or Akita; &&P<0.01 vs HG+IL-17A+Ad-GFP (or HG+IL-17A) or Akita+Ad-GFP; NS (no significance) vs HG+IL-17A or Akita. Ad-Act1-shRNA, adenoviral vector that expressed short hairpin RNA targeting Act1; Ad-GFP, adenoviral vector that expressed green fluorescent protein; HG, high glucose; IL, interleukin; NF-κB nuclear factor-κB; PBS, phosphate-buffered saline; TRAF6, tumor necrosis factor receptor-associated factor-6.
In the retinas of the Akita mice, the Act1 and TRAF6 expression and p65 phosphorylation were increased compared with the WT mice (Figure 2b). IL-17A injection in the vitreous cavity of the Akita mice further increased the retinal Act1 and TRAF6 expression and p65 phosphorylation, whereas Ad-Act1-shRNA intravitreal treatment in the Akita mice reduced the retinal Act1 and TRAF6 expression and p65 phosphorylation compared with the Ad-GFP intravitreal injection (Figure 2b). In a comparison of the effects between two doses of IL-17A, the high dose of IL-17A (40 ng per 2 μl) was stronger than the low dose of IL-17A (8 ng per 2 μl) (P<0.05 for Act1 expression; P<0.01 for TRAF6 expression and p65 phosphorylation).
IL-17A exacerbates HG-induced Müller cell activation and dysfunction via Act1/IKK signaling
In the HG condition, the retinal Müller cells increased the GFAP expression and production (Figures 3a and b), VEGF expression and secretion (Figures 3a and c) and glutamate production (Figure 3d); however, they decreased the GS and EAAT1 expression (Figure 3a). IL-17A treatment exacerbated the HG-induced Müller cell activation and functional impairment (Figures 3a–d). Act1 gene silencing using Ad-Act1-shRNA or IKK activity inhibition using Wedel in Müller cells abrogated the IL-17A deleterious effects (Figures 3a–d). Ad-Act1-shRNA or Wedel treatment alone (no IL-17A treatment) also reduced the HG-induced changes in the expression, production and/or secretion of GFAP, VEGF, GS, EAAT1 and glutamate in the Müller cells (Figures 3a–d).

IL-17A exacerbates HG-induced Müller cell activation and dysfunction via Act1/IKK signaling. (a) Expression of proteins related to Müller cell activation (GFAP) and function (VEGF, GS and EAAT1) in cultured Müller cells. (b) GFAP content in cultured Müller cell lysates assessed via ELISA. (c) VEGF concentration in cultured Müller cell supernatants assessed via ELISA. (d) Glutamate content in Müller cell lysates measured by HPLC. The treatment time periods of IL-17A, Ad-Act1-shRNA and the IKK inhibitor Wedel were the same as described in Figure 2. The cutline at the lower left of this figure is shared by (a–d). **P<0.01 vs control; +P<0.05, ++P<0.01 vs HG; &&P<0.01 vs HG+IL-17A+Ad-GFP or HG+IL-17A; NS (no significance) vs HG+IL-17A. Ad-Act1-shRNA, adenoviral vector that expressed short hairpin RNA targeting Act1; Ad-GFP, adenoviral vector that expressed green fluorescent protein; ELISA, enzyme-linked immunosorbent assay; EAAT1, excitatory amino acid transporter-1; GFAP, glial fibrillary acidic protein; GS, glutamine synthetase; HG, high glucose; HPLC, high-performance liquid chromatography; IL, interleukin; VEGF, vascular endothelial growth factor.
IL-17A aggravates in vivo retinal Müller cell activation and dysfunction in Akita mice via Act1 signaling
In the retinas of the Akita mice, the GFAP and VEGF expression was upregulated; however, the GS and EAAT1 expression was downregulated compared with the WT mice (Figure 4a). IL-17A intravitreal administration in the Akita mice exacerbated the retinal expression changes, whereas Ad-Act1-shRNA intravitreal treatment in the Akita mice alleviated these retinal expression changes compared with the Ad-GFP intravitreal injection (Figure 4a). Similarly, the increased glutamate content in the retinas of the Akita mice was further increased by IL-17A intravitreal administration; however, it was reduced by Ad-Act1-shRNA intravitreal treatment (Figure 4b).

IL-17A aggravates in vivo Müller cell activation and dysfunction in Akita mouse retina via Act1 signaling. At 3 months after the onset of diabetes, Akita mice were injected in the vitreous cavity with IL-17A, Ad-Act1-shRNA (or Ad-GFP as a control) or anti-IL-17A mAb. At 2 days after the intravitreal injections of IL-17A or anti-IL-17A mAb or 2 weeks after the intravitreal injection of Ad-Act1-shRNA, the tests were performed. (a) Expression of proteins related to Müller cell activation (GFAP) and function (VEGF, GS and EAAT1) in the retina. (b, d) Glutamate content in the retina assessed via HPLC. (c) Immunofluorescent staining on retinal cross-sections indicates a coexpression of GS that labels Müller cells and VEGF. **P<0.01 vs WT mice; +P<0.05, ++P<0.01 vs Akita or Akita+PBS; &&P<0.01 vs Akita+Ad-GFP; NS (no significance) vs Akita. Ad-Act1-shRNA, adenoviral vector that expressed short hairpin RNA targeting Act1; Ad-GFP, adenoviral vector that expressed green fluorescent protein; EAAT1, excitatory amino acid transporter-1; GCL, ganglion cell layer; GFAP, glial fibrillary acidic protein; GS, glutamine synthetase; HPLC, high-performance liquid chromatography; IL, interleukin; INL, inner nuclear layer; mAb, monoclonal antibody; ONL, outer nuclear layer; PBS, phosphate-buffered saline; VEGF, vascular endothelial growth factor.
To demonstrate that the retinal changes were linked to Müller cells, we examined the coexpression of GS and VEGF in the retina. Fluorescent immunohistochemistry indicated that the expression changes in GS and VEGF in the retina by the treatments were similar to the changes demonstrated by western blot analysis (Figure 4c). Notably, the Akita mouse retina had an increased coexpression of GS and VEGF compared with the WT mouse retina. This increase was further enhanced by IL-17A intravitreal administration; however, it was reduced by Ad-Act1-shRNA intravitreal treatment (Figure 4c).
To confirm that IL-17A promoted Müller cell functional impairment, we assessed the effect of a blocking antibody to IL-17A. Intravitreal injection of anti-IL-17A mAb (2 or 10 μg per 2 μl) in the Akita mice ameliorated the diabetes-induced increase in the retinal glutamate content (Figure 4d), and the high dose of anti-IL-17A mAb (10 μg per 2 μl) had a larger effect than the low dose of anti-IL-17A mAb (2 μg per 2 μl) (P<0.05). As a control of the intravitreal administration, a PBS intravitreal injection did not significantly affect the diabetes-induced retinal glutamate accumulation (Figure 4d).
IL-17A exacerbates Akita mouse BRB breakdown via Act1 signaling
BRB breakdown was determined by retinal vascular leukostasis and vascular leakage. In the retinas of the Akita mice, the number of vascular adherent leukocytes per retina was increased (Figure 5a) and the FITC-labeled dextran leakage was enhanced (Figure 5b) compared with the WT mice. These retinal vascular lesions in the Akita mice were aggravated by IL-17A intravitreal injection; however, they were alleviated by intravitreal treatment with Ad-Act1-shRNA or anti-IL-17A mAb (Figures 5a and b). Moreover, the increase in the vascular adherent leukocyte number was greater with the high dose of IL-17A (40 ng per 2 μl) than with the low dose of IL-17A (8 ng per 2 μl) (P<0.01). As a control of Ad-Act1-shRNA, Ad-GFP intravitreal injection in the Akita mice did not significantly alter the diabetes-induced retinal vascular leukostasis and vascular leakage (Figures 5a and b).

IL-17A exacerbates BRB breakdown in Akita mice via Act1 signaling. (a) Retinal flat-mounted images indicate vascular leukostasis. The treatment time periods of IL-17A, Ad-Act1-shRNA and anti-IL-17A mAb were the same as described in Figure 4. The adherent leukocytes labeled by FITC-concanavalin A within retinal vessels were counted in each retina. **P<0.01 vs wild-type (WT) mice; ++P<0.01 vs Akita mice; &&P<0.01 vs Akita+Ad-GFP; NS (no significance) vs Akita. (b) Retinal angiography indicates retinal vascular leakage. The retinal angiography was performed 2 weeks after the intravitreal treatments with IL-17A, Ad-Act1-shRNA or anti-IL-17A mAb. The arrows indicate the vascular FITC–dextran exudation in the flat-mounted retinas. Ad-Act1-shRNA, adenoviral vector that expressed short hairpin RNA targeting Act1; Ad-GFP, adenoviral vector that expressed green fluorescent protein; BRB, blood–retinal barrier; FITC, fluorescein isothiocyanate; IL, interleukin; mAb, monoclonal antibody.
IL-17A increases retinal ganglion cell apoptosis in Akita mice via Act1 signaling
The numbers of both NeuN/TUNEL double-positive cells and NeuN/active-caspase-3 double-positive cells were increased in the retinal ganglion cell layer of the Akita mice compared with that of the WT mice (Figures 6a–c). IL-17A intravitreal administration in the Akita mice further increased the numbers of both NeuN/TUNEL double-stained cells and NeuN/active-caspase-3 double-labeled cells in the retinal ganglion cell layer (Figures 6a–c). These effects of increasing apoptotic neuronal cell numbers by IL-17A were stronger at the high dose (40 ng per 2 μl) than at the low dose (8 ng per 2 μl) (P<0.01 or P<0.05). In contrast, Ad-Act1-shRNA intravitreal treatment in the Akita mice reduced the apoptotic neuronal cell numbers compared with Ad-GFP intravitreal treatment (Figures 6a–c).

IL-17A increases retinal ganglion cell apoptosis in Akita mice via Act1 signaling. The treatment time periods of IL-17A, Ad-Act1-shRNA and anti-IL-17A mAb were the same as described in Figure 4. (a) Immunofluorescent staining of retinal cross-sections indicates NeuN/TUNEL double-positive cells in the GCL. (b) Immunofluorescent staining of retinal cross-sections indicates NeuN/Active-Cas (Active-caspase-3) double-positive cells in the GCL. The arrows in (a, b) indicate representative double-labeled cells. (c) Statistical histogram for (a, b). The data were obtained as described in the Materials and methods. (d) Caspase-3 activity in the retina that was determined by the ratio of active-caspase-3 (Active-Cas) and pro-caspase-3 (Pro-Cas). **P<0.01 vs wild-type (WT) mice; ++P<0.01 vs Akita mice or Akita+PBS; &&P<0.01 vs Akita+Ad-GFP; NS (no significance) vs Akita. Ad-Act1-shRNA, adenoviral vector that expressed short hairpin RNA targeting Act1; Ad-GFP, adenoviral vector that expressed green fluorescent protein; GCL, ganglion cell layer; IL, interleukin; mAb, monoclonal antibody; TUNEL, TdT-mediated dUTP nick end labeling.
In addition, the caspase-3 activity in the diabetic retinas was upregulated, and this upregulation was attenuated by an intravitreal injection of anti-IL-17A mAb (Figure 6d). This effect of reducing the caspase-3 activity by anti-IL-17A mAb was increased at its high dose (10 μg per 2 μl) compared with the low dose (2 μg per 2 μl) (P<0.05).
Discussion
In the present study, we demonstrated that diabetes increased the retinal expression of IL-17A and IL-17RA as well as the production of IL-17A, suggesting that the proinflammatory cytokine IL-17A is involved in DR. Fluorescent immunohistochemistry indicated that IL-17A and IL-17RA were expressed by Müller cells in contrast to ganglion cells in diabetic retinas. Additional in vitro evidence demonstrated that HG induced IL-17A expression in and secretion from retinal Müller cells. These findings suggest that retinal Müller cells are a source and target of IL-17A in DR. IL-17A is often deemed a specific proinflammatory cytokine of Th17 cells. The retina does not have well-developed lymphatic drainage;39 thus, the ocular tissue is considered immunoprivileged. Several recent studies suggest that Th17 cells infiltrate from the circulation into the eye across the retinal vascular endothelium to participate in the inflammatory response during degeneration of the retinal pigment epithelium40 and in autoimmune uveitis.41 Here, we provide novel evidence that demonstrates IL-17A is also produced by retinal Müller cells in DR.
Müller cells play a critical role in DR occurrence and development. Müller cell activation, demonstrated by increased GFAP expression, is a sign of human early DR pathology.42 Müller cells are also a major source of retinal VEGF in diabetic mice, and Müller cell-derived VEGF plays an essential and causative role in retinal inflammation and vascular leakage in DR.27 In addition, retinal glutamate accumulation, as a result of Müller cell downregulation of GS and EAAT1 in DR, leads to excitotoxicity, particularly to retinal neurons.43, 44 These disorders in the expression, production and/or secretion of GFAP, VEGF, GS, EAAT1 and glutamate by Müller cells in DR were demonstrated in this study. Importantly, exogenously administered IL-17A exacerbated Müller cell activation and dysfunction in both in vitro and in vivo diabetic models, whereas blocking IL-17A ameliorated diabetes-induced retinal glutamate accumulation. These findings suggest that Müller cells are actively involved in the inflammatory response to IL-17A, in which this proinflammatory cytokine promotes DR occurrence and development.
The complete IL-17A signaling cascade is far from completely defined; however, it has been reported that a signaling transduction pathway that involves Act1/TRAF6/IKK/NF-κB is activated after IL-17A binds to IL-17RA.17, 45 In the current study, HG and diabetes induced IL-17RA, Act1 and TRAF6 expression as well as p65 phosphorylation in Müller cells and retinal tissue, respectively. These findings suggest that during DR, the inflammatory signaling cascade IL-17RA/Act1/TRAF6/IKK/NF-κB is activated most likely in Müller cells. IL-17A further enhanced the activation of this inflammatory signaling cascade, and Act1 knockdown or IKK inhibition blocked the signaling activation by IL-17A in Müller cells. These findings demonstrate that this inflammatory signaling pathway mediates the IL-17A proinflammatory property in Müller cells. Moreover, exacerbated Müller cell activation and functional impairment in vitro by IL-17A were abolished by Act1 silencing or IKK inhibition. Similarly, in the retinal tissue of Akita mice, Müller cell activation and functional abnormalities were reduced by Act1 knockdown or IL-17A inhibition. Thus, an inhibitor that inhibits the activation of the inflammatory signaling cascade IL-17RA/Act1/TRAF6/IKK/NF-κB represents a promising, novel therapeutic strategy for DR.
BRB deterioration, which is characterized by reduced blood flow, leukostasis, increased vascular permeability and pathological angiogenesis, is a cardinal manifestation of DR.46, 47 We demonstrated that IL-17A intravitreal administration exacerbated diabetes-induced retinal vascular leukostasis and vascular leakage, whereas Act1 knockdown or IL-17A inhibition alleviated the retinal vascular damages. These findings demonstrate a crucial contribution of IL-17A to DR-like pathology. BRB is composed of both inner and outer barriers. The outer BRB is formed by the retinal pigment epithelium cell layer, and the inner BRB comprises the microvascular endothelium that lines the retinal microvasculature.48 The tight junctions located between these cells mediate the highly selective diffusion of molecules from the blood to the retina. IL-17A compromises ARPE-19 (retinal pigment epithelium cell line) monolayer barrier function in association with a disrupted distribution of the junction proteins zonula occludens-1 and occludin.49 These findings suggest a mechanism that underlies IL-17A injury to BRB. In addition, abnormal Müller cell function has been linked to BRB breakdown.50 Thus, we propose that the IL-17A effect of accelerating BRB disruption may be implemented by promoting Müller cell functional impairment in the DR process.
To date, DR is widely accepted as a neurovascular disease rather than a sole vascular disease.44 The apoptosis of retinal neurons is reported in DR patients and mice.51, 52 In retinal neurons, ganglion cells represent the best studied neurons with respect to the effects of diabetes.53 In this study, diabetes-induced retinal caspase-3 activity was inhibited by IL-17A-blockade, suggesting that IL-17A mediates retinal apoptosis in the DR process. Additional evidence indicated that diabetes-induced retinal ganglion cell apoptosis was increased by IL-17A administration and decreased by Act1 knockdown. Retinal ganglion cells did not express IL-17RA, similar to brain neurons;54 thus, IL-17A damage to retinal ganglion cells is not a direct action on the cells. We explain that the decrease in glutamate recycling in dysfunctional Müller cells in response to IL-17A is a critical cause of neuronal death in DR. A Müller cell abnormality has been associated with retinal neuronal dysfunction and death.50 Moreover, Müller cells make contact with both neuronal somata and processes in the entire retina; they thus play key roles in the homeostasis and metabolism of retinal neurons and maintain a healthy and properly functioning retina.55 Collectively, the retinal neuronal apoptosis in DR may be an outcome of Müller cell functional impairment.
In conclusion, diabetes induces IL-17A production and IL-17RA/Act1/TRAF6/IKK/NF-κB signaling activation in retinal Müller cells. Exogenously administered IL-17A exacerbates diabetes-induced Müller cell activation and dysfunction as well as BRB disruption and retinal neuronal apoptosis, whereas blocking IL-17A ameliorates the pathological alterations of the retina. In addition, inhibition of this inflammatory signaling cascade using Act1 knockdown or an IKK inhibitor alleviates the DR-like pathology. These findings demonstrate that IL-17A production and Act1 signaling activation in retinal Müller cells during DR represent an inflammatory response that accelerates DR progression.
References
Cheung N, Mitchell P, Wong TY . Diabetic retinopathy. Lancet 2010; 376: 124–136.
Semeraro F, Cancarini A, dell'Omo R, Rezzola S, Romano MR, Costagliola C . Diabetic retinopathy: vascular and inflammatory disease. J Diabetes Res 2015; 2015: 582060.
Gariano RF, Gardner TW . Retinal angiogenesis in development and disease. Nature 2005; 438: 960–966.
Wang LL, Chen H, Huang K, Zheng L . Elevated histone acetylations in Muller cells contribute to inflammation: a novel inhibitory effect of minocycline. Glia 2012; 60: 1896–1905.
Abcouwer SF . Angiogenic factors and cytokines in diabetic retinopathy. J Clin Cell Immunol 2013 S1 011–022.
Demircan N, Safran BG, Soylu M, Ozcan AA, Sizmaz S . Determination of vitreous interleukin-1 (IL-1) and tumour necrosis factor (TNF) levels in proliferative diabetic retinopathy. Eye 2006; 20: 1366–1369.
Suzuki Y, Nakazawa M, Suzuki K, Yamazaki H, Miyagawa Y . Expression profiles of cytokines and chemokines in vitreous fluid in diabetic retinopathy and central retinal vein occlusion. Jpn J Ophthalmol 2011; 55: 256–263.
Adamis AP, Berman AJ . Immunological mechanisms in the pathogenesis of diabetic retinopathy. Semin Immunopathol 2008; 30: 65–84.
Hazra S, Rasheed A, Bhatwadekar A, Wang X, Shaw LC, Patel M et al. Liver X receptor modulates diabetic retinopathy outcome in a mouse model of streptozotocin-induced diabetes. Diabetes 2012; 61: 3270–3279.
Huang H, Gandhi JK, Zhong X, Wei Y, Gong J, Duh EJ et al. TNFalpha is required for late BRB breakdown in diabetic retinopathy, and its inhibition prevents leukostasis and protects vessels and neurons from apoptosis. Invest Ophthalmol Vis Sci 2011; 52: 1336–1344.
Shen X, Xie B, Cheng Y, Jiao Q, Zhong Y . Effect of pigment epithelium derived factor on the expression of glutamine synthetase in early phase of experimental diabetic retinopathy. Ocul Immunol Inflamm 2011; 19: 246–254.
Aggarwal S, Gurney AL . IL-17: prototype member of an emerging cytokine family. J Leukoc Biol 2002; 71: 1–8.
Gaffen SL . Recent advances in the IL-17 cytokine family. Curr Opin Immunol 2011; 23: 613–619.
Semeran K, Pawlowski P, Lisowski L, Szczepaniak I, Wojtowicz J, Lawicki S et al. Plasma levels of IL-17, VEGF, and adrenomedullin and S-cone dysfunction of the retina in children and adolescents without signs of retinopathy and with varied duration of diabetes. Mediators Inflamm 2013; 2013: 274726.
Marwaha AK, Crome SQ, Panagiotopoulos C, Berg KB, Qin H, Ouyang Q et al. Cutting edge: increased IL-17-secreting T cells in children with new-onset type 1 diabetes. J Immunol 2010; 185: 3814–3818.
Hang H, Yuan S, Yang Q, Yuan D, Liu Q . Multiplex bead array assay of plasma cytokines in type 2 diabetes mellitus with diabetic retinopathy. Mol Vis 2014; 20: 1137–1145.
Gu C, Wu L, Li X . IL-17 family: cytokines, receptors and signaling. Cytokine 2013; 64: 477–485.
Hinz M, Scheidereit C . The IkappaB kinase complex in NF-kappaB regulation and beyond. EMBO Rep 2014; 15: 46–61.
Chong RS, Martin KR . Glial cell interactions and glaucoma. Curr Opin Ophthalmol 2015; 26: 73–77.
Reichenbach A, Bringmann A . New functions of Muller cells. Glia 2013; 61: 651–678.
Bringmann A, Wiedemann P . Muller glial cells in retinal disease. Ophthalmologica 2012; 227: 1–19.
Fu S, Dong S, Zhu M, Sherry DM, Wang C, You Z et al. Muller glia are a major cellular source of survival signals for retinal neurons in diabetes. Diabetes 2015; 64: 3554–3563.
Vacca O, Darche M, Schaffer DV, Flannery JG, Sahel JA, Rendon A et al. AAV-mediated gene delivery in Dp71-null mouse model with compromised barriers. Glia 2014; 62: 468–476.
Barber AJ, Antonetti DA, Gardner TW . Altered expression of retinal occludin and glial fibrillary acidic protein in experimental diabetes. The Penn State Retina Research Group. Invest Ophthalmol Vis Sci 2000; 41: 3561–3568.
Carrasco E, Hernandez C, Miralles A, Huguet P, Farres J, Simo R . Lower somatostatin expression is an early event in diabetic retinopathy and is associated with retinal neurodegeneration. Diabetes Care 2007; 30: 2902–2908.
Garcia-Ramirez M, Hernandez C, Villarroel M, Canals F, Alonso MA, Fortuny R et al. Interphotoreceptor retinoid-binding protein (IRBP) is downregulated at early stages of diabetic retinopathy. Diabetologia 2009; 52: 2633–2641.
Wang J, Xu X, Elliott MH, Zhu M, Le YZ . Muller cell-derived VEGF is essential for diabetes-induced retinal inflammation and vascular leakage. Diabetes 2010; 59: 2297–2305.
Gu L, Xu H, Wang F, Xu G, Sinha D, Wang J et al. Erythropoietin exerts a neuroprotective function against glutamate neurotoxicity in experimental diabetic retina. Invest Ophthalmol Vis Sci 2014; 55: 8208–8222.
Zeng K, Xu H, Chen K, Zhu J, Zhou Y, Zhang Q et al. Effects of taurine on glutamate uptake and degradation in Muller cells under diabetic conditions via antioxidant mechanism. Mol Cell Neurosci 2010; 45: 192–199.
Robinson R, Barathi VA, Chaurasia SS, Wong TY, Kern TS . Update on animal models of diabetic retinopathy: from molecular approaches to mice and higher mammals. Dis Model Mech 2012; 5: 444–456.
Lai AK, Lo AC . Animal models of diabetic retinopathy: summary and comparison. J Diabetes Res 2013; 2013: 106594.
Li J, Wang JJ, Yu Q, Chen K, Mahadev K, Zhang SX . Inhibition of reactive oxygen species by lovastatin downregulates vascular endothelial growth factor expression and ameliorates blood-retinal barrier breakdown in db/db mice: role of NADPH oxidase 4. Diabetes 2010; 59: 1528–1538.
Zhong Y, Li J, Chen Y, Wang JJ, Ratan R, Zhang SX . Activation of endoplasmic reticulum stress by hyperglycemia is essential for Muller cell-derived inflammatory cytokine production in diabetes. Diabetes 2012; 61: 492–504.
Kumar A, Shamsuddin N . Retinal Muller glia initiate innate response to infectious stimuli via toll-like receptor signaling. PLoS ONE 2012; 7: e29830.
Mysona BA, Al-Gayyar MM, Matragoon S, Abdelsaid MA, El-Azab MF, Saragovi HU et al. Modulation of p75(NTR) prevents diabetes- and proNGF-induced retinal inflammation and blood-retina barrier breakdown in mice and rats. Diabetologia 2013; 56: 2329–2339.
Zhang Y, Zhang J, Wang Q, Lei X, Chu Q, Xu GT et al. Intravitreal injection of exendin-4 analogue protects retinal cells in early diabetic rats. Invest Ophthalmol Vis Sci 2011; 52: 278–285.
Han Z, Guo J, Conley SM, Naash MI . Retinal angiogenesis in the Ins2(Akita) mouse model of diabetic retinopathy. Invest Ophthalmol Vis Sci 2013; 54: 574–584.
Harada C, Guo X, Namekata K, Kimura A, Nakamura K, Tanaka K et al. Glia- and neuron-specific functions of TrkB signalling during retinal degeneration and regeneration. Nat Commun 2011; 2: 189.
Schroedl F, Brehmer A, Neuhuber WL, Kruse FE, May CA, Cursiefen C . The normal human choroid is endowed with a significant number of lymphatic vessel endothelial hyaluronate receptor 1 (LYVE-1)-positive macrophages. Invest Ophthalmol Vis Sci 2008; 49: 5222–5229.
Zhao Z, Xu P, Jie Z, Zuo Y, Yu B, Soong L et al. gammadelta T cells as a major source of IL-17 production during age-dependent RPE degeneration. Invest Ophthalmol Vis Sci 2014; 55: 6580–6589.
Bharadwaj AS, Schewitz-Bowers LP, Wei L, Lee RW, Smith JR . Intercellular adhesion molecule 1 mediates migration of Th1 and Th17 cells across human retinal vascular endothelium. Invest Ophthalmol Vis Sci 2013; 54: 6917–6925.
Vujosevic S, Micera A, Bini S, Berton M, Esposito G, Midena E . Aqueous humor biomarkers of Muller cell activation in diabetic eyes. Invest Ophthalmol Vis Sci 2015; 56: 3913–3918.
Hernandez C, Bogdanov P, Corraliza L, Garcia-Ramirez M, Sola-Adell C, Arranz JA et al. Topical administration of GLP-1 receptor agonists prevents retinal neurodegeneration in experimental diabetes. Diabetes 2016; 65: 172–187.
Jindal V . Neurodegeneration as a primary change and role of neuroprotection in diabetic retinopathy. Mol Neurobiol 2015; 51: 878–884.
Zhong B, Liu X, Wang X, Chang SH, Liu X, Wang A et al. Negative regulation of IL-17-mediated signaling and inflammation by the ubiquitin-specific protease USP25. Nat Immunol 2012; 13: 1110–1117.
Cerani A, Tetreault N, Menard C, Lapalme E, Patel C, Sitaras N et al. Neuron-derived semaphorin 3A is an early inducer of vascular permeability in diabetic retinopathy via neuropilin-1. Cell Metab 2013; 18: 505–518.
Tang J, Kern TS . Inflammation in diabetic retinopathy. Prog Retin Eye Res 2011; 30: 343–358.
Campbell M, Humphries P . The blood-retina barrier: tight junctions and barrier modulation. Adv Exp Med Biol 2012; 763: 70–84.
Chen Y, Yang P, Li F, Kijlstra A . The effects of Th17 cytokines on the inflammatory mediator production and barrier function of ARPE-19 cells. PLoS ONE 2011; 6: e18139.
Fletcher EL, Downie LE, Ly A, Ward MM, Batcha AH, Puthussery T et al. A review of the role of glial cells in understanding retinal disease. Clin Exp Optom 2008; 91: 67–77.
Mohr S, Xi X, Tang J, Kern TS . Caspase activation in retinas of diabetic and galactosemic mice and diabetic patients. Diabetes 2002; 51: 1172–1179.
Park HY, Kim JH, Park CK . Neuronal cell death in the inner retina and the influence of vascular endothelial growth factor inhibition in a diabetic rat model. Am J Pathol 2014; 184: 1752–1762.
Kern TS, Barber AJ . Retinal ganglion cells in diabetes. J Physiol 2008; 586: 4401–4408.
Kawanokuchi J, Shimizu K, Nitta A, Yamada K, Mizuno T, Takeuchi H et al. Production and functions of IL-17 in microglia. J Neuroimmunol 2008; 194: 54–61.
Fernandez-Sanchez L, Lax P, Campello L, Pinilla I, Cuenca N . Astrocytes and Muller cell alterations during retinal degeneration in a transgenic rat model of retinitis pigmentosa. Front Cell Neurosci 2015; 9: 484.
Acknowledgements
This study was supported by the National Basic Research Program of China (973 Program, No. 2013CB967500 and No. 2011CB510200), the National Natural Science Foundation of China (No. 81170855), the Jiangsu Provincial Special Program of Medical Science of China (BL2014089) and the Six Talent Peak Project in Jiangsu Province of China (2013-WSW-015).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Qiu, AW., Bian, Z., Mao, PA. et al. IL-17A exacerbates diabetic retinopathy by impairing Müller cell function via Act1 signaling. Exp Mol Med 48, e280 (2016). https://doi.org/10.1038/emm.2016.117
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2016.117
This article is cited by
-
Glial cell alterations in diabetes-induced neurodegeneration
Cellular and Molecular Life Sciences (2024)
-
Network pharmacology mechanism of Scutellarin to inhibit RGC pyroptosis in diabetic retinopathy
Scientific Reports (2023)
-
The role of the adaptive immune system and T cell dysfunction in neurodegenerative diseases
Journal of Neuroinflammation (2022)
-
Neutrophil breaching of the blood vessel pericyte layer during diapedesis requires mast cell-derived IL-17A
Nature Communications (2022)
-
Inhibition of CD40-TRAF6-dependent inflammatory activity halts the onset of diabetic retinopathy in streptozotocin-diabetic mice
Nutrition & Diabetes (2022)
